Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124


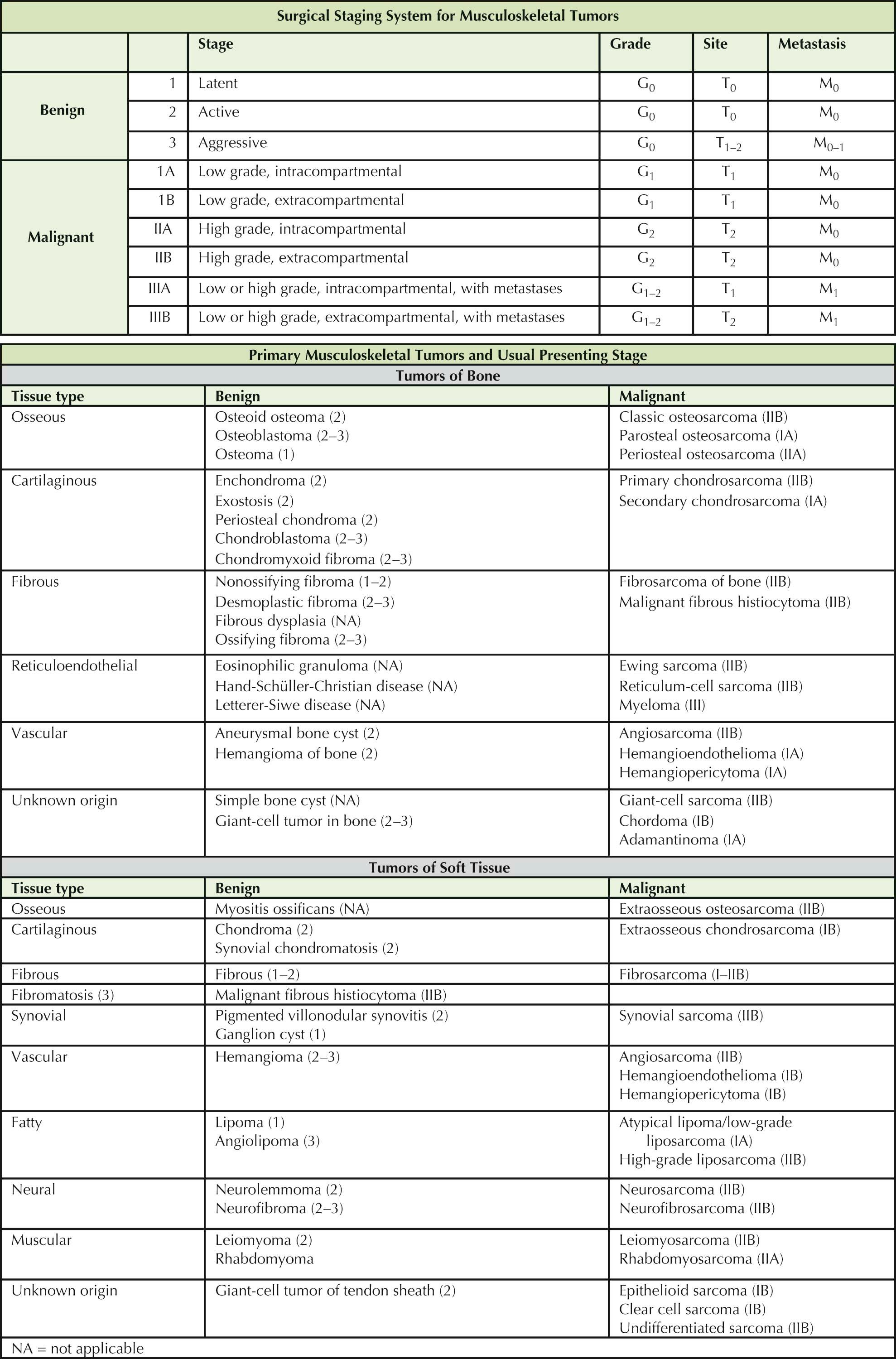
An understanding of the tumors of the musculoskeletal system requires a thorough knowledge of clinical presentation, natural history, staging characteristics, histopathology, and response to treatment of these tumors. Common bone tumors include myeloma, lymphoma, and metastases from primary breast, lung, kidney, thyroid, and prostate cancers; and these are, in fact, the most common malignant bone lesions in patients older than 40 years of age. Much less common are primary lesions of bone. Osteosarcoma is the most common primary bone tumor, followed by Ewing sarcoma and chondrosarcoma. A classification of primary (musculoskeletal origin) benign and malignant tumors of bone and soft tissue is presented in the table.
Staging studies may be needed to determine the local extent of lesions or distant metastases and histologic diagnosis of the disease.
Radionuclide bone scans are performed to assess multiple sites of involvement, the extent of local intraosseous involvement not apparent on radiographs, and tumor activity.
Computed tomography (CT) of the lesion is employed for determining bone destruction, intrinsic density of the lesion, and cortical extension. Metastases of primary musculoskeletal sarcomas are most commonly found in the lungs. Chest radiographs and chest CT are used for chest surveillance to provide information about metastatic disease. The role of combined positron emission tomography and computed tomography (PET/CT) is unclear, but it has been most useful in delineation of neurofibromas from sarcomas in patients with neurofibromatosis.
Magnetic resonance imaging (MRI) delineates the precise location and extent of tumor involvement. Specifically, MRI is the primary study used to delineate compartmental involvement, intra-articular involvement, and proximity of neurovascular structures; MRI provides superior resolution and sensitivity in depicting abnormalities and is particularly useful in determining the extent of soft tissue lesions and the subtle involvement of the bone marrow in bone lesions.
Staging. The staging system for musculoskeletal tumors shown in Table 6-1 represents an assessment of the surgical grade, local extent of disease, and presence or absence of metastases. It is based on the stratification and interrelationship of these three factors and is used to predict the prognosis, response to surgical treatment, risk of local recurrence, and metastatic potential.
The surgical grade (G) reflects a tumor's aggressiveness based on its histologic pattern: benign (G 0 ), low-grade malignant (G 1 ), and high-grade malignant (G 2 ).
The local extent of disease (T) defines the primary lesion as intracapsular (T 0 , surrounded by an intact capsule of fibrous tissue or reactive bone); extracapsular but intracompartmental (T 1 , remaining within an intraosseous, intrafascial or intramuscular, periosteal, or paraosseous compartment or potential compartment); and extracapsular and extracompartmental (T 2 , extending beyond its compartment of origin or arising within incompletely bounded spaces, such as the popliteal fossa, axilla, or groin).
The designation M indicates the presence or absence of metastases: no known metastases (M 0 ) or metastases present (M 1 ).

Osteoid osteoma is a benign osseous tumor that occurs primarily in adolescents and less often in children and young adults. The most common site is the proximal femur; and although osteoid osteoma typically involves the diaphysis of long bones, it may also occur in the foot (talus, navicular, or calcaneus) and in the posterior elements of the spine, where it can lead to a secondary scoliosis. The typical presenting symptom is well-localized pain that is most severe at night and is relieved by aspirin and other prostaglandin inhibitors. The lesion typically has a nidus, but this can be difficult to locate on radiograph. CT or MRI can be helpful in locating the nidus and increasing the certainty of diagnosis.
Diagnostic Studies. An intense bony reaction to a small nidus is the radiographic hallmark of osteoid osteoma. Radiographs reveal an oval radiolucent nidus only 3 to 5 mm in diameter and surrounded by a disproportionately large, dense reactive zone. Although usually located in the cortex, a nidus may occur in the subperiosteal and endosteal regions. CT scans at 5-mm intervals are used to confirm a cortical nidus and to help direct the therapeutic approach. The bone scan usually shows increased radioisotope uptake.
The radiographic differential diagnosis includes osteomyelitis (chronic sclerosing osteomyelitis), Brodie's abscess, and stress fracture.
Histologic examination reveals a nidus composed of thick, vascular bars of osteoblastic tissue surrounded by a thin zone of vascular fibrous tissue, in turn surrounded by a dense shell, or margin, of mature reactive cortical bone. The histologic differential diagnosis primarily includes osteoblastoma (see Plate 6-3 ). Although osteoblastoma is similar to osteoid osteoma in many respects, it is usually larger and has some subtle but distinct histologic differences. Distinguishing osteosarcoma (see Plates 6-15 and 6-16 ) from the small osteoblastic nidus of osteoid osteoma is rarely problematic.
Treatment/Prognosis. Although osteoid osteoma may eventually resolve spontaneously, with spontaneous ossification and the subsequent relief of pain, most patients prefer not to wait for resolution, owing to severe pain. Percutaneous radiofrequency ablation of the osteoid osteomas can be performed with up to 90% success and can be done under local anesthesia with CT scan guidance; thus, this is the current initial treatment of choice. When the nidus is located in a low-stress area such as the metaphysis, en bloc excision with a surrounding small block of reactive bone can be performed but has increased morbidity relative to percutaneous ablation. Alternatively, the overlying margin of reactive bone may be shaved until the nidus is visible as a cherry-red spot; this spot then can be removed with curettage. Although intracapsular curettage is associated with a higher recurrence rate than other types of excision, it minimizes the risk of postoperative fracture in high-stress areas such as the femoral neck. Most recurrences result from fragmentation of the lesion, partial excision, or inaccurate localization of the lesion in an inaccessible place.
After radiofrequency ablation, complete excision, or even spontaneous resolution, prognosis is excellent. No cases of malignant transformation have been reported.

Osteoblastoma is a rare benign osseous tumor. Because of its many similarities to osteoid osteoma but larger size (generally > 2 cm), it was at one time called giant osteoid osteoma. Osteoblastoma can be seen in slightly older people than osteoid osteoma. Osteoid osteoma is most common in older adolescents, and osteoblastoma is most common in older adolescents and young adults. There are also important clinical, radiographic, and histologic differences between osteoid osteoma and osteoblastoma. For example, osteoblastoma does not cause well-localized night pain that is relieved by aspirin and it occurs more often in the posterior elements of the vertebrae (transverse and spinous processes and pedicles) and jaw.
Diagnostic Studies. Plain radiographs show a relatively radiolucent, bone-forming (osteoblastic) lesion that is osteolytic and often has an aneurysmal, or blown-out, appearance. It is surrounded by a thin margin of reactive bone that frequently extends into adjacent soft tissue. The intense bony reaction around osteoid osteoma does not occur with osteoblastoma.
The radiographic differential diagnosis includes osteoid osteoma (see Plate 6-2 ), aneurysmal bone cyst (see Plate 6-11 ), eosinophilic granuloma (see Plate 6-10 ), giant cell tumor of bone (see Plate 6-13 ), and osteosarcoma (see Plates 6-15 and 6-16 ).
Bone scans demonstrate an intense radioisotope uptake, but MRI helps to determine the surgical approach.
The histologic appearance of osteoblastoma is quite similar to that of osteoid osteoma but the former has a more prominent vascular component. In addition, osteoblastoma has more stromal tissue and giant cells and broader osteoid seams than osteoid osteoma. These characteristics also distinguish osteoblastoma from osteosarcoma. However, the distinction of osteoblastoma from osteosarcoma is not always clear. For example, osteoblastomas often contain scattered mitotic figures and a proliferation of immature osteoblasts.
Treatment/Prognosis. Osteoblastomas can recur after intracapsular procedures, which are usually done to preserve joint function particularly in the spine.
Some cases of malignant transformation of osteoblastoma into osteosarcoma have been reported, and it is not clear whether this is due to the difficulty in initial diagnosis or genuine transformation. Regardless, patients with osteoblastoma must be observed not only for recurrence but also for malignant transformation.

Enchondroma is a benign primarily asymptomatic cartilaginous tumor of the intramedullary aspect of bone usually in the metaphysis (see Plate 6-4 ).
Enchondromas can occur anywhere in the skeleton. In very rare cases, a benign enchondroma undergoes malignant transformation into secondary chondrosarcoma (see Plate 6-17 ). However, aggressive appearing lesions in the hands are still nearly always benign. The uncommon occurrence of multiple lesions is known as enchondromatosis, or Ollier disease, and these patients more commonly have malignant degeneration of their enchondromas so they require surveillance of their lesions (see Plate 6-17 ).
Diagnostic Studies. Radiographs show a central radiolucent lesion with a well-defined but minimally thickened bony margin. During the active phase in adolescence, the lesion may slowly enlarge. In the inactive, or latent, phase in adulthood, the cartilaginous tissue may calcify in a diffuse punctate or stippled configuration. These calcifications sometimes appear on the radiograph as subtle “smoke ring” or “ring and arc” images. As the lesion matures, it develops a more reactive margin.
Bone scans can demonstrate radioisotope uptake. CT or MRI may be used to assess cortical erosion, intralesional calcification, and exact location. CT is also useful to rule out a soft tissue mass, which is indicative of chondrosarcoma. There are parathyroid hormone receptor-1 (PTHR1) mutations in about 10% of patients with enchondromatosis.
Biopsy is usually not needed to confirm the diagnosis of enchondroma, because its cartilaginous nature is evident radiographically and it is not destructive of the cortex, although these benign lesions can scallop the inside of the cortex. It can be difficult to distinguish between an active benign enchondroma and a low-grade malignant chondrosarcoma on the basis of histologic examination alone. Therefore, if there is radiographic evidence of full-thickness cortical destruction (not just endosteal scalloping from within) or MRI appearance of a soft tissue mass, this may provide evidence that the lesion is a chondrosarcoma.
Treatment/Prognosis. Asymptomatic solitary enchondromas are presumed benign and require only follow-up if pain develops or there is an increase in size. If solitary or multiple enchondromas become symptomatic and begin to enlarge, staging studies with radiographs (and if still indeterminate then MRI) are indicated to rule out malignancy.
The prognosis for benign enchondroma is excellent. The lesion usually becomes latent in adulthood, and less than 1% of asymptomatic solitary enchondromas become malignant. In enchondromatosis, however, the risk of malignant transformation is much greater (approximately 10% of cases).

Periosteal chondroma is a cartilaginous dysplasia that arises beneath the perichondrium and produces a broad-based, hemispheric cartilaginous mass that bulges from the cortex into the soft tissues. Typically seen in adolescents and young adults, the tumor manifests as a painless mass on the surface of a major long bone, most commonly the lateral cortex of the proximal humerus just proximal to the insertion of the deltoid muscle (see Plate 6-5 ).
Diagnostic Studies. Periosteal chondromas appear on radiographs as a radiolucent oval or oblong defect visualized as a crater-like deformity of the periphery of the cortex. The lesion is underlined by a thin, distinct cortical reaction. The tumor has little or no calcification. MRI can identify the density of the cartilage as high on T2-weighted sequences.
The radiographic differential diagnosis includes osteocartilaginous exostosis in younger patients (see Plate 6-6 ), juxtacortical chondrosarcoma (see Plate 6-17 ), as well as juxtacortical (parosteal and periosteal) osteosarcomas (see Plate 6-16 ).
On gross examination, the lesion has the consistency of mature cartilage without evidence of ossification or calcification. Histologic examination reveals hypercellular cartilage, and nuclear atypia may be present.
Treatment/Prognosis. Periosteal chondromas are active benign stage 2 tumors that require marginal excision to prevent recurrence. Because of their peripheral location and their cortical rim, this can usually be achieved without bone grafting or extensive reconstruction. There is a risk of recurrence after excision. Sarcomatous transformation has not been documented, and rare reports of such transformation probably reflect the resemblance of periosteal chondroma to juxtacortical chondrosarcoma.

Osteocartilaginous exostosis (osteochondroma) is a common developmental dysplasia of the peripheral growth plate that results in a lobulated outgrowth of cartilage and bone from the metaphysis. The classic presentation is a hard excrescence of bone capped by a thin zone of proliferating cartilage. An exostosis may develop in any bone but is usually seen in the long bones. The most common locations are the proximal or distal femur, proximal humerus, proximal tibia, pelvis, and scapula. The tumor develops in adolescence and continues to enlarge during skeletal growth.
The initial clinical sign is a hard, painless mass fixed on the bone. Symptoms, when present, are usually due to irritation of the overlying soft tissues that may or may not be associated with a fluid-filled bursa. Variations in the fluid content in the bursa create a fluctuating, palpable mass.
In most patients, the lesion is solitary; however, polyostotic tumors occur in a small subset of patients. These multiple hereditary exostoses produce significant deformities, such as short stature and angular deformities. In multiple hereditary exostoses as with enchondromatosis there is an increased incidence of malignant degeneration.
Diagnostic Studies. The radiographic appearance of an exostosis is either a flat, sessile lesion or a pedunculated (stalklike) cortical-sharing process. The lesions are called cortical sharing because they share the same cortex as the adjacent normal cortical bone. It is usually a well-defined metaphyseal projection of bone with mottled density; the cartilaginous cap displays irregular areas of calcification. The radiographic hallmark is the blending of the tumor into the underlying metaphysis. Although a diagnosis is seldom problematic, the presence of a painful and enlarging lesion in an adult may necessitate staging studies to assess the risk of secondary malignant transformation to chondrosarcoma (see Plate 6-17 ).
Evidence of malignant transformation includes confirmation by CT or MRI of a soft tissue mass with cortical destruction and a sudden increase in size of the mass and pain. The radiographic differential diagnosis is periosteal chondroma (see Plate 6-5 ) and parosteal osteosarcoma (see Plate 6-16 ). In an adult, a symptomatic “exostosis” that increases in size could be a malignancy, although benign osteochondromas may develop a reactive bursa that increases in size, simulating tumor growth. About 70% of patients with multiple exostoses have germline EXT1 or EXT2 mutations.
Exostoses on gross examination reveal an overlying cartilaginous cap with no growth plate but separated from the underlying bone by an irregular, chalky white line (zone of calcification). The mass of trabecular bone that makes up the main portion of the exostosis merges into the underlying normal trabecular bone of the metaphysis.
On microscopic examination, the cartilaginous cap does not have the tidemark characteristic of articular cartilage.
Treatment/Prognosis. Marginal excision of an active exostosis, including the cartilaginous cap and the overlying perichondrium, minimizes the risk of recurrence. The deep bony base has minimal activity and may be removed piecemeal (in more than one piece). The prognosis for a solitary exostosis is excellent (<5% recurrence after marginal excision). The risk of sarcomatous transformation in solitary exostosis is about 1%, but in multiple hereditary exostoses, this risk approaches 10%.

Chondroblastoma is a painful, benign cartilaginous tumor that arises during adolescence in the epiphysis close to the joint surface and most commonly in the proximal humerus, proximal tibia, or distal femur.
Diagnostic Studies. The radiographic hallmark is an epiphyseal radiolucent lesion with fine punctate calcifications suggestive of a cartilaginous lesion. The tumor is usually bordered by a well-defined margin of reactive bone. Chondroblastomas are more epiphyseally based than the more common epimetaphyseal giant cell tumor. The radiographic differential diagnosis includes aneurysmal bone cyst (see Plate 6-11 ) and giant cell tumor of bone (see Plate 6-13 ). Inflammatory lesions such as osteoarthritic cysts also are epiphyseal but are nearly always in older patients, and chondroblastomas are generally in young patients frequently with open growth plates. Giant cell tumors are more common in young adults with closed growth plates.
Histologic examination reveals the characteristic “cobblestone” or “chicken-wire” pattern: areas of round, plump chondroblasts (the stones) are enmeshed in a sparse chondroid matrix (the mortar between the stones). Clumps of giant cells are seen in areas that contain primarily a spindle cell stroma. Unique to chondroblastoma is the fine microscopic pattern of calcifications, usually in a “chicken-wire” arrangement, in and around the islands of cartilage.
Treatment/Prognosis. Curettage is the treatment of choice of chondroblastomas. Curettage near the growth plate must be undertaken with great care in the younger patient to prevent late angular deformities, and curettage near the articular cartilage must be undertaken with great care in all patients.
The prognosis for chondroblastoma is good; most are active stage 2 tumors; however, there is a significant risk of recurrence after curettage similar to that of giant cell tumor.
Chondromyxoid fibroma is a rare painless, benign cartilaginous lesion of bone. It occurs in adolescents, most often in the metaphyses of major long bones. This lesion most often presents as an active stage 2 lesion and is not known to undergo malignant transformation.
Diagnostic Studies. Radiographs reveal an eccentric metaphyseal radiolucent defect with no evidence of the usual calcification of a cartilaginous tumor. The radiographic differential diagnosis includes nonossifying fibroma (see Plate 6-9 ) and aneurysmal bone cyst (see Plate 6-11 ).
Histologic examination shows immature myxoid cartilage with stellate chondrocytes enmeshed in lightly staining myxomatous chondroid matrix. Intertwined throughout the lesion are strands of benign fibrous tissue and small multinucleated giant cells.
Treatment. Curettage carries a low risk of recurrence in well-encapsulated stage 2 lesions, but the size of the defect often necessitates bone grafting (see Plate 6-30 ).

Fibrous dysplasia is a developmental abnormality of bone that results in a haphazard mixture of immature fibrous tissue and small fragments of immature trabecular bone. The lesions may occur in one bone (monostotic) or in many (polyostotic), and they are sometimes accompanied by precocious puberty and café-au-lait pigmentation in females, making up the triad known as McCune-Albright syndrome.
Monostotic lesions generally occur in the proximal femur, proximal tibia, mandible, and ribs. Polyostotic disease, which usually presents earlier, may be unilateral or widespread, affecting long bones, hands, feet, facial bones, and pelvis. Extensive involvement of the proximal femur results in the distinctive “shepherd's crook” deformity that is characteristic of fibrous dysplasia.
The result of this dysplastic process is a weakened bone that becomes deformed by normal stress or sustains frequent pathologic fractures. Painful stress fractures are especially common in the proximal femur. Although dysplastic bone heals at a normal rate after fracture, the resulting callus is also dysplastic, and the disease persists.
Diagnostic Studies. The classic radiographic feature of fibrous dysplasia is a hazy, radiolucent, or “ground-glass” pattern resulting from the defective mineralization of immature dysplastic bone; it is usually strikingly different from the radiographic appearance of normal bone, calcified cartilage, or soft tissue. A small monostotic lesion may be difficult to distinguish from other benign lesions, but extensive polyostotic involvement is likely to produce the characteristic ground-glass density and significant deformity. The lesion is primarily diaphyseal or metaphyseal and has been described as a “long lesion in a long bone.”
Bone scans demonstrate intense radioisotope uptake, and CT scans help visualize the ground-glass density of the lesion. MRI can delineate the extent of involvement and show the characteristic fat or cystic nature of some of the lesions. Fat in the fibrous dysplasia lesions may be present on MRI and is reassuring that the lesion is benign.
The typical histologic pattern is an irregular collection of small pieces of immature bone within a matrix of fibrous tissue. The overall histologic appearance has been likened to that of alphabet soup. The immature trabeculae are not lined with osteoblasts (as in ossifying fibroma), do not contain cement lines, and are obviously not aligned according to stress. The fibrous stroma is loosely arranged and immature, replacing the normal marrow. A variable degree of capillary vasculature is seen within the stroma.
The histologic differential diagnosis includes osteoblastoma (see Plate 6-3 ), osteosarcoma (see Plate 6-15 ), ossifying fibroma, hyperparathyroidism, and Paget disease of bone. Specific GNAS activating (gain-of-function) mutations have been described in fibrous dysplasia and McCune-Albright syndrome.
Treatment. Because more dysplastic bone usually forms and pain does not predictably resolve after curettage, the goal of management should be conservative and prevention of deformity and fracture. This is best accomplished using cortical bone allografts (taken from the fibula), which minimally remodel after incorporation. Treatment methods for severe bony involvement are reconstruction with joint replacements or internal metallic fixation.

Nonossifying fibroma (fibrous cortical defect, metaphyseal fibrous defect) is the most common bone lesion. It results from a developmental defect of periosteal cortical bone that leads to a failure of ossification during the normal growth period. This lesion, of fibrous origin, typically develops in childhood with a slightly higher incidence in males. Although usually asymptomatic, nonossifying fibroma can be an active lesion that persists or enlarges throughout childhood. When the tumor occupies a large portion of the bone, fracture can occur. Fractures through nonossifying fibromas will usually heal, but open reduction may be necessary depending on the location. With skeletal maturation, a nonossifying fibroma becomes latent and ossifies.
Diagnostic Studies. The lesion commonly develops in the metaphysis of the distal femur or distal tibia and is eccentrically located, usually within or adjacent to the cortex. Radiographs reveal a well-marginated radiolucent zone, with distinct trabeculation producing a multilocular appearance. In addition, nonossifying fibromas usually cause benign cortical thinning, or erosion. The radiographic pattern is usually diagnostic, and further staging studies are seldom indicated.
Histologic features include a combination of dense collagen arranged in a storiform pattern, a scattering of small, multinucleated giant cells, hemosiderin, and lipid-filled histiocytes. The tissue does not contain the degree of hemorrhage and necrosis or the number of giant cells seen in the giant cell tumor of bone. Although the tissue may resemble the lining tissue of an aneurysmal bone cyst, the lesion does not have a large, central, blood-filled cavity.
Treatment. Reassurance and watchful waiting is usually sufficient except with fracture when closed or open reduction and immobilization with bone grafting is necessary.
Desmoplastic fibroma (desmoid tumor) is a rare intraosseous fibroma that typically develops as an aggressive stage 3 tumor. It occurs primarily in young adults but may occur at any age. The long bones—particularly the tibia and the fibula—are the most common sites, although it may occur throughout the skeleton. Its behavior corresponds to that of its soft tissue counterpart, aggressive fibromatosis (see Plate 6-23 ).
Diagnostic Studies. Radiographs show a centrally located metaphyseal or diaphyseal lesion, poorly or incompletely contained by a thin margin of reactive bone, which frequently has a trabeculated appearance. It may remain within the bone for some time, surrounded by a thin cortical shell, but eventually it extends through the cortex into the soft tissues. The radiographic hallmark is a loculated intraosseous lesion that stimulates very little bony reaction. The radiographic differential diagnosis includes giant cell tumor of bone (see Plate 6-13 ) and fibrosarcoma of bone (see Plate 6-18 ). The most characteristic feature is the contrast between the relatively benign findings of the radiographic studies and the tumor's fairly aggressive clinical behavior.
The lesion is composed of dense, white, fibrous tissue with a rubbery consistency and is easily removed with curettage. The histologic features of dense, irregularly arranged bundles of collagen with an occasional spindle cell closely resemble fibromatosis. Mitoses, vascularity, and necrosis are unusual microscopic findings. The histologic differential diagnosis usually involves low-grade fibrosarcoma of bone.
Treatment. Excision with a wide margin results in local control of the lesion. Recurrence after curettage is frequent, with invasion of the soft tissue.

Eosinophilic granuloma belongs to a group of disorders characterized by lesions that occur as a result of metabolic defects in the reticuloendothelial system. This group of disorders, called histiocytosis X, also includes Hand-Schüller-Christian disease and Letterer-Siwe disease. The clinical and radiographic manifestations of histiocytosis X may resemble those of a malignancy, and these lesions can be multifocal, but their clinical course differs from that of malignant reticuloendothelial tumors such as lymphoma, Ewing sarcoma (see Plate 6-19 ), leukemia, Hodgkin disease, and multiple myeloma (see Plate 6-20 ).
Eosinophilic granuloma usually occurs as a solitary, symptomatic lesion in children younger than 20 years of age but can occur as multiple tumors at any age and at any site. Low-grade fever, elevated erythrocyte sedimentation rate, and mild peripheral eosinophilia are occasional associated findings. The skull, supra-acetabular region of the pelvis, and diaphysis of the femur are the usual sites of involvement, although any bone can be affected.
Diagnostic Studies. On radiographs, a small lesion may resemble the punched-out radiolucent lesion of multiple myeloma. Involvement of a vertebral body produces the pathognomonic radiographic appearance of vertebra plana (“coin-on-edge” flattened vertebra) after vertebral collapse. A lesion that occurs in the diaphysis or metaphysis of a limb appears as an oval radiolucent area. Because of its variable radiographic characteristics, eosinophilic granuloma has been called the great imitator.
Bone scans are used to document the presence of multiple lesions (<10% of cases); an intense radioisotope uptake indicates an active lesion. MRI is helpful in delineating the lesion, especially in the spine and hip.
Gross examination of an active lesion reveals soft, vascular, granulomatous tissue covered with a mature bony capsule that may be runny and liquefied. Histologic characteristics are a mixture of Langerhans histiocytes, eosinophils, and occasional giant cells with little background stroma. Birbeck granules can also be seen. The same histologic pattern is seen in each of the forms of histiocytosis X, but the multisystem Hand-Schüller-Christian and Letterer-Siwe diseases have significant morbidity and mortality and are sometimes treated with chemotherapy.
Treatment/Prognosis. The curettage performed to obtain tissue for diagnosis is usually adequate treatment. When the defect is large, bone grafts may be needed to prevent fracture (see Plate 6-30 ). In a small percentage of patients with a solitary lesion, the visceral manifestations of Hand-Schüller-Christian disease can eventually develop and follow-up or a bone scan should be performed to rule out multifocal disease.

The aneurysmal bone cyst is a tumor-like proliferation of vascular tissue that forms a lining around a blood-filled cystic lesion. Occurring most commonly in adolescents and young adults, it develops in the metaphyseal region of long bones, pelvis, or vertebral body, sometimes secondary to another benign or malignant bone lesion.
Diagnostic Studies. A radiolucent lesion with a ballooned expansion of the bone cortices (“finger in balloon”) is the radiographic hallmark of an aneurysmal bone cyst. Although some lesions appear to have an aggressive, expansile quality, they remain contained by a thin rim of reactive periosteal bone. In children, these benign lesions seldom penetrate the articular surface of a joint or the growth plate; therefore, evidence of growth plate penetration by an aneurysmal bone cyst indicates the need for careful staging studies to rule out malignancy. The radiographic differential diagnosis includes simple bone cyst (see Plate 6-12 ), giant cell tumor of bone (see Plate 6-13 ), telangiectatic sarcoma, and angiosarcoma (see Plate 6-27 ).
Bone scans show intense radioisotope uptake in the margin of the lesion. MRI and CT are used to depict the precise anatomic extent and density of the lesion, and especially the thin, limiting margin of reactive bone (which is not well visualized on plain radiographs). A fluid-fluid level seen on the MR image generally confirms the diagnosis and represents the separation between the red cells and serum in the bloody cavity. Other tumors can, however, have fluid-fluid levels and aneurysmal bone cyst–like qualities.
Histologically, the proliferative lining tissue of an aneurysmal bone cyst is often difficult to distinguish from that of a giant cell tumor of bone. It contains a mixture of benign stromal tissue, giant cells, and large amounts of hemosiderin. The tissue usually contains large vascular lacunae lined with giant cells and filled with clotted blood that resembles cranberry sauce. When these histologic features also occur in other lesions (e.g., chondroblastoma, osteoblastoma, osteosarcoma, eosinophilic granuloma, nonossifying fibroma) they are called secondary aneurysmal bone cysts. Because of the preponderance of giant cells, many aneurysmal bone cysts are initially thought to be giant cell tumors of bone, and there is a continuum between giant cell tumors of bone and aneurysmal bone cysts.
Treatment/Prognosis. The majority of active aneurysmal bone cysts are treated with curettage and bone grafting; the recurrence rate, however, is 20% to 30%.
After incision of an active cyst, an alarming amount of bleeding may occur until the lining is completely removed. After complete excision of the cyst this bleeding generally decreases. Occasionally, however, there may still be brisk bleeding emanating directly from the bony wall, which can be controlled with coagulation. Curettage may be augmented with cementation with methyl methacrylate, which appears to reduce the risk of recurrence or bone graft, which allows for healing and for a recurrence to be visualized earlier. Lesions in the pelvis and spine have a higher risk of recurrence, where complete surgical exposure and removal are more difficult. Nevertheless, the prognosis for primary aneurysmal bone cyst is excellent and recurrences can usually be managed with more aggressive curettage or excision.

The simple (unicameral) bone cyst is a membrane-lined cavity containing a clear yellow fluid. Occurring most often in children 4 to 10 years of age, the cysts have a predilection for the metaphysis of long bones, particularly the proximal humerus (50% of cases), proximal femur, and proximal tibia. The lesions remain asymptomatic unless complicated by fracture. They enlarge during skeletal growth and become inactive, or latent, after skeletal maturity.
Active cysts usually develop in patients younger than 10 years of age. Typically, the cyst abuts the growth plate and occupies most of the metaphysis; it is expansile with a thin cortical shell, continues to enlarge during observation, and is commonly associated with fracture. The cyst is considered active as it arises and grows adjacent to the growth plate. With growth, it is left behind and becomes increasingly separated from the growth plate and at this point is considered latent.
Latent cysts are usually seen in patients older than 12 years of age. They have a thicker bony wall than active lesions. They remain static or diminish in size, show evidence of healing or ossification, and are less likely to result in fracture.
Diagnostic Studies. Radiographs show a central, well-marginated radiolucent defect in the metaphysis, with a symmetric appearance, usually without bony septations or loculations. The metaphysis is expanded, with marked cortical thinning that predisposes to fracture. After fracture, the eggshell-thin fragments are often displaced into the cystic cavity, thus creating the “falling leaf” sign ( Plate 6-12 ).
The radiographic differential diagnosis is usually limited to fibrous dysplasia (see Plate 6-8 ) and aneurysmal bone cyst (see Plate 6-11 ). In some cases, the even radiolucency of the cyst may be difficult to distinguish from the ground-glass density of fibrous dysplasia. However, monostotic fibrous dysplasia is usually eccentric rather than central and diaphyseal rather than metaphyseal; in addition, the periosteal reaction is greater in fibrous dysplasia than in a simple bone cyst. An aneurysmal bone cyst (and even telangiectatic sarcoma) may also appear as a large radiolucent lesion, making it occasionally difficult to distinguish from a simple cyst.
Staging studies are indicated only when radiographs are difficult to interpret. Bone scans show an increased radioisotope uptake around the margin of the cyst, in contrast to the uniform uptake in fibrous dysplasia. The finding of straw-colored fluid on needle aspiration confirms the diagnosis of a simple cyst.
Histologic examination of an active cyst reveals a fibrous membrane lining a thin margin of reactive bone. The inner wall of the margin deep to the membrane is often covered with a network of osteoclasts. Between the membrane and the osteoclastic activity is a layer of areolar tissue containing fibroblastic and multinucleated giant cells. Latent cysts have a thicker membrane with little underlying osteoclastic activity, fewer giant cells, and more reactive bone.
Treatment/Prognosis. Simple cysts are treated with curettage and bone grafting; recurrence is high for active cysts (50%) and low for latent cysts (10%). Treatment with injection of corticosteroid is based on the theory that corticosteroids stabilize the mesothelial lining and induce healing of the cyst. Cyst fluid is aspirated with sterile percutaneous needle technique; then 80 to 200 mg of methylprednisolone acetate is infused into the cavity. In more than half of patients studied, the cyst healed after this technique, although multiple injections frequently were required. If the lesion fractures, there is a small chance that the fracture can heal and the lesion resolve.

Giant cell tumor of bone (osteoclastoma) is a common benign but locally aggressive lesion. It occurs chiefly in persons between 20 and 40 years of age. The lesion is typically located in the epiphysis and metaphysis of the distal femur or proximal tibia. Other common sites are the distal radius, proximal humerus, distal tibia, and sacrum. The tumor usually enlarges to occupy most of the epiphysis and adjacent metaphysis, penetrating and eroding the subchondral bone and even invading the articular cartilage. Patients report a deep, persistent intraosseous pain that mimics an internal derangement of the knee. A pathologic fracture or reactive knee effusion is the initial symptom in about one third of patients; in a small number (<1%), the tumor undergoes pulmonary metastases that are generally but not always benign.
Diagnostic Studies. Radiographs reveal a large radiolucent lesion surrounded by a distinct margin of reactive bone. Cortical thinning, endosteal erosion, and trabecularization, or bony septation, of the cavity are associated findings. CT or MRI discloses the extent of involvement and bony margination. Lesions extend to with 1.5 cm of the adjacent joint space and articular cartilage. A chest radiograph should be performed in all patients with giant cell tumor to rule out metastases.
Gross examination reveals a soft, friable, reddish brown neoplastic tissue with the consistency of a wet sponge. Some areas are gelatinous or fatty, and some are aneurysmal and cavitated. Histologic features include multinucleated giant cells, a proliferative stroma with vesiculated nuclei, areas of aneurysmal tissue, areas of necrosis, reactive peripheral bone, and occasional mitotic figures and intravascular tumor plugs in venous sinuses.
Treatment. The size and stage of the tumor determine the type of treatment. For stage 1 or 2 lesions, curettage is often combined with bone grafting or cementation. Giant cell tumors of bone have a recurrence rate between 5% and 30%; Cementation and adjuvant treatment with phenol or liquid nitrogen may decrease the recurrence rate. For recurrent lesions or articular destruction, extensive joint reconstruction or joint replacement may be required.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here
