Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

The thyroid gland is located in the anterior neck, lying like a small bow tie across the front of the trachea. In adults, the normal thyroid weighs ~20 g. It is composed of left and right lobes and a small connecting branch, or isthmus.
The thyroid gland possesses many features unique among endocrine glands, not the least of which is that it is the only endocrine gland common to both sexes that can be easily seen and palpated in the course of a routine clinical examination. At the biochemical level, the thyroid hormones are the only ones that require an essential trace element, iodine, for the production of active hormone. Another unusual feature of thyroid hormone physiology is that the hormone is stored in an extracellular site within a highly proteinaceous material called thyroid colloid. The major protein within this material is thyroglobulin, which contains—as part of its primary structure—the thyroid hormones tetraiodothyronine (T 4 or thyroxine) and triiodothyronine (T 3 ). Thyroglobulin, the sequestered prohormone, is entirely surrounded by thyroid follicular cells, which are responsible for the synthesis of thyroid hormones ( Fig. 49-1 ).
The physiological actions of thyroid hormones also display several unique aspects. Although derived from a large protein (i.e., thyroglobulin), T 4 and T 3 are not peptides. Unlike peptide hormones or hormones derived from single amino acids (e.g., catecholamines), no cell membrane receptors exist for T 4 or T 3 . Instead, like the steroid hormones, thyroid hormones act principally by binding to nuclear receptors (see pp. 71–72 ) and regulate the transcription of cell proteins. T 4 and T 3 act on multiple tissues and are essential for normal development, growth, and metabolism.
The C cells (parafollicular cells) of the thyroid, which are not part of the follicular unit, synthesize another hormone, calcitonin (see Fig. 49-1 ). Calcitonin may play a role in Ca 2+ and phosphate homeostasis, and we discuss its physiology on pages 1067–1068 .
The structures of T 4 and T 3 , the two active thyroid hormones, are shown in Figure 49-2 . T 3 is far more active than T 4 . Also shown is reverse T 3 (rT 3 ), which has no known biological activity. It has two iodines on its outer benzyl ring, rather than two on its inner ring, as is the case for T 3 . All three compounds derive from the ether linkage of a tyrosine molecule to the benzyl group of a second tyrosine molecule; one or two iodine atoms are attached to each benzyl group. The bottom panel of Figure 49-2 shows T 4 as part of the thyroglobulin molecule.
; rT 3 is present in approximately equal molar amounts with T 3 . However, rT 3 is essentially devoid of biological activity. As shown in the bottom panel, T 4 is part of the peptide backbone of the thyroglobulin molecule, as are T 3 and rT 3 . Cleavage of the two indicated peptide bonds would release T 4 .")
The synthesis of thyroid hormones begins with the trapping of iodine by the thyroid gland. Iodine is essential for the formation of thyroid hormones. It exists in nature as a trace element in soil and is incorporated into many foods. The iodide anion (I − ) is rapidly absorbed by the gastrointestinal (GI) tract and is actively taken up via a specialized Na/I cotransporter or symporter ( NIS or SLC5A5; see p. 122 ), located at the basolateral membrane (i.e., facing the blood) of the thyroid follicular cell ( Fig. 49-3 ). NIS is a 65-kDa integral membrane protein that is believed to have 13 membrane-spanning segments. NIS moves two Na + and one I − into the follicular cell against the I − electrochemical gradient, fueled by the energy of the Na + electrochemical gradient (see pp. 120–123 ). Several other anions (e.g., perchlorate, pertechnetate, and thiocyanate) can compete with I − for uptake by the thyroid. Iodide leaves the follicular cell and enters the lumen of the follicle across the apical membrane. The Cl-I exchanger pendrin (SLC26A4; see p. 125 ) ![]() N49-1 is present on the apical membrane, where it contributes to I − secretion. Mutations in this protein can lead to a congenital syndrome typically characterized by a large thyroid gland (goiter) and hearing loss. The thyroid enlarges because of deficient I − delivery to the follicular colloid, just as it would with an I − -deficient diet ( Box 49-1 ).
N49-1 is present on the apical membrane, where it contributes to I − secretion. Mutations in this protein can lead to a congenital syndrome typically characterized by a large thyroid gland (goiter) and hearing loss. The thyroid enlarges because of deficient I − delivery to the follicular colloid, just as it would with an I − -deficient diet ( Box 49-1 ).

In areas where soil is relatively iodine deficient, human iodine deficiency is common. Because seawater and seafood contain large amounts of iodide, iodine deficiency is more common in inland areas, particularly in locales that rely on locally grown foods. For example, in inland areas of South America along the Andes Mountains, in central Africa, and in highland regions of Southeast Asia, iodine deficiency is common. In the early 1900s, investigators first recognized that iodide is present in high concentrations in the thyroid and that iodine deficiency promotes goiter formation. These observations led to efforts to supplement dietary iodine. Iodine deficiency causes thyroid hormone deficiency. The pituitary responds to this deficit by increasing the synthesis of TSH (see p. 1014 ), which in turn increases the activity of the iodine-trapping mechanism in the follicular cell in an effort to overcome the deficiency. The increased TSH also exerts a trophic effect that increases the size of the thyroid gland. If this trophic effect persists for sufficient time, the result is an iodine-deficiency goiter. Goiter is the generic term for an enlarged thyroid. If this effort at compensation is not successful (i.e., if insufficient thyroid hormone levels persist), the person will develop signs and symptoms of goitrous hypothyroidism. When iodine deficiency occurs at critical developmental times in infancy, the effects on the CNS are particularly devastating and produce the syndrome known as cretinism (see p. 1013 ). Persons so affected have a characteristic facial appearance and body habitus, as well as severe mental retardation. Dietary supplementation of iodine in salt and bread has all but eliminated iodine deficiency in North America. In many nations, especially in mountainous and landlocked regions of developing countries, iodine deficiency remains a major cause of preventable illness.
Pendred syndrome —the combination of congenital hypothyroidism and acoustic nerve deafness—is caused by a mutation in SLC26A4, which encodes an anion exchanger called pendrin. The syndrome accounts for as much as 10% of the cases of inherited deafness and is also often associated with a goiter.
In parallel with the secretion of I − into the follicle lumen, the follicular cell secretes thyroglobulin (Tg) into the lumen; Tg contains the tyrosyl groups to which the I − will ultimately attach. The Tg molecule is a glycoprotein synthesized within the follicular cell and exported to the follicular lumen via the secretory pathway (see p. 28 ). Tg is a very large protein (>600 kDa), and it accounts for approximately half of the protein content of the thyroid gland. It has relatively few tyrosyl residues (~100 per molecule of Tg), and only a few of these (<20) are subject to iodination. The secretory vesicles that contain Tg also carry the enzyme thyroid peroxidase (TPO) —an integral membrane protein, with the catalytic domain facing the vesicle lumen. As the secretory vesicles fuse with the apical membrane, the catalytic domain faces the follicular lumen and catalyzes the oxidation of I − to I 0 . This reaction requires H 2 O, provided by another apical membrane protein, dual-oxidase 2 (DUOX2). As the Tg is entering the lumen of the thyroid follicle by the process of exocytosis, its tyrosyl groups react with I 0 .
One or two oxidized iodine atoms incorporate selectively into specific tyrosyl residues of Tg. TPO in the presence of H 2 O 2 catalyzes the coupling of two iodinated tyrosyl residues within the Tg molecule to form a single iodothyronine as well as a remnant dehydroalanine. Both remain as part of the primary structure of the iodinated Tg until it is later degraded inside the follicular cell. This coupling of two tyrosines, catalyzed by TPO, does not occur unless they are iodinated. Because only a few tyrosyl groups become iodinated, something specific about the structure of the protein near these residues probably facilitates both iodination and conjugation. The thyroid hormones, although still part of the Tg molecule, are stored as colloid in the thyroid follicle.
Thyroid hormones, attached to Tg in the follicular lumen (see Fig. 49-1 ), remain inactive until the iodinated Tg is hydrolyzed (see Fig. 49-3 ). Before this proteolysis can begin, the follicular cells must resorb Tg from the follicular lumen by fluid-phase endocytosis (see pp. 41–42 ). As the endocytic vesicle containing the colloid droplet moves from the apical toward the basolateral membrane, it fuses with lysosomes to form a lysoendosome. Inside this vesicle, lysosomal enzymes hydrolyze the Tg and form T 4 and T 3 , as well as diiodothyronine (DIT) and monoiodothyronine (MIT). The vesicle releases both T 4 and T 3 near the basolateral membrane, and these substances exit the cell into the blood by an unknown mechanism. Approximately 90% of the thyroid hormone secreted by the thyroid is released as T 4 , and 10% is released as T 3 . The thyroid releases very little rT 3 into the blood. As discussed in the next section, nonthyroidal tissues metabolize the T 4 released by the thyroid into T 3 and rT 3 . Approximately three fourths of circulating T 3 arises from the peripheral conversion of T 4 , which occurs principally in the liver and kidneys.
In the circulation, both T 4 and T 3 are highly bound to plasma proteins. Thyroid-binding globulin (TBG), albumin, and transthyretin (TTR) account for most of this binding. The affinity of these binding proteins is sufficiently high that, for T 4 , >99.98% of the hormone circulates tightly bound to protein. T 3 is bound only slightly less: ~99.5% is protein bound. Because the free or unbound hormone in the circulation is responsible for the actions of the thyroid hormones on their target tissues, the large amount of bound hormone has considerably confounded our ability to use simple measurements of the total amount of either T 4 or T 3 in the plasma to provide a reliable index of the adequacy of thyroid hormone secretion. For example, the amount of TBG in the serum can change substantially in different physiological states. Pregnancy, oral estrogen therapy, hepatitis, and chronic heroin abuse can all elevate the amount of TBG and hence the total concentration of T 4 and T 3 . Decreased levels of TBG, associated with diminished concentration of total T 4 and T 3 , can accompany steroid usage and nephrotic syndrome. However, despite the marked increases or decreases in the amounts of circulating TBG, the concentrations of free T 4 and T 3 do not change in the aforementioned cases. Box 49-2 indicates how one can calculate levels of free T 4 or T 3 , knowing the concentration of TBG and the concentration of total T 4 or total T 3 .
Most of the T 4 and T 3 in the serum is bound to proteins, the most important of which is TBG. For the binding of T 4 to TBG, the reaction is
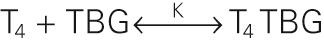
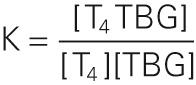
The binding constant K is ~2 × 10 10 M −1 for T 4 . The comparable binding constant for T 3 is ~5 × 10 8 M −1 . Approximately one third of TBG's binding sites are occupied by T 4 . Therefore, we have all the information we need to compute the concentration of free T 4 :
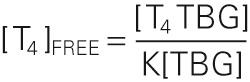
A reasonable value for [T 4 TBG] would be 100 nM, and for [TBG], 250 nM. Thus,

Because the bound T 4 in this example is 100 nM, and the free T 4 is only 20 pM, we can conclude that only ~0.02% of the total T 4 in the plasma is free. Because 99.98% of the total T 4 in the plasma is bound, moderate fluctuations in the rate of T 4 release from the thyroid have only tiny effects on the level of free T 4 . To simplify, we have not included the minor contribution of albumin and TTR in this sample calculation.
The liver makes each of the thyroid-binding proteins. TBG is a 54-kDa glycoprotein consisting of 450 amino acids. It has the highest affinity for T 4 and T 3 and is responsible for most of the thyroid-binding capacity in the plasma. The extensive binding of thyroid hormones to plasma proteins serves several functions. It provides a large buffer pool of thyroid hormones in the circulation, so that the active concentrations of hormone in the circulation change very little on a minute-to-minute basis. The binding to plasma proteins markedly prolongs the half-lives of both T 4 and T 3 . T 4 has a half-life of 8 days, and T 3 , of ~24 hours; each is longer than the half-life of steroid or peptide hormones. Finally, because much of the T 3 in the circulation is formed by the conversion of T 4 to T 3 in extrathyroidal tissues, the presence of a large pool of T 4 in the plasma provides a reserve of prohormone available for synthesis of T 3 . This reserve may be of particular importance because T 3 is responsible for most of the biological activity of thyroid hormones.
The thyroid synthesizes and stores much more T 4 than T 3, and this is reflected in the ~10 : 1 ratio of T 4 to T 3 secreted by the thyroid. However, certain tissues in the body have the capacity to selectively deiodinate T 4 , thereby producing either T 3 or rT 3 . T 3 and rT 3 can each be further deiodinated to various DITs and MITs ( Fig. 49-4 ); both DITs and MITs are biologically inactive. Both iodine atoms on the inner ring, and at least one iodine atom on the outer ring, appear essential for biological activity. Similarly, the loss of the amino group renders T 4 or T 3 inactive. The importance of the peripheral deiodination of T 4 to T 3 can be readily appreciated from the observation that persons whose thyroids have been removed have normal circulating concentrations of T 3 when they receive oral T 4 supplementation.
 remove I from the outer benzyl ring, whereas the 5/3-monodeiodinase (type 3; orange arrows ) removes I from the inner benzyl ring. Thus, the action of the 5′/3′-monodeiodinases on T 4 yields T 3 , whereas the action of the 5/3-monodeiodinase yields rT 3 . Sequential deiodination yields T 0 (thyronine).")
Inasmuch as T 3 is biologically much more active than the far more abundant T 4 , the regulated conversion of T 4 to T 3 in peripheral tissues—as well the conversion of T 4 and T 3 to inactive metabolites—assumes considerable importance. These conversions are under the control of three deiodinases. Two deiodinases are 5′/3′-deiodinases that remove an I from the outer ring and thereby convert T 4 to T 3 (see Fig. 49-4 ). The first of these 5′/3′-deiodinases— type 1 deiodinase —is present at high concentrations in the liver, kidneys, skeletal muscle, and thyroid. It appears to be responsible for generating most of the T 3 that reaches the circulation. The second 5′/3′-deiodinase— type 2 deiodinase —is found predominantly in the pituitary, central nervous system (CNS), and placenta, and is involved in supplying those tissues with T 3 by local generation from plasma-derived T 4 . As shown below, the type 2 enzyme in the pituitary is of particular importance because the T 3 that is generated there is responsible for the feedback inhibition of the release of thyrotropin (or thyroid-stimulating hormone, TSH ).
A third 5/3-deiodinase— type 3 deiodinase —removes an I from the inner ring, thereby converting T 4 to the inactive rT 3 . Because the 3′ and 5′ positions in T 4 are equivalent stereochemically, removing either of these by type 1 or type 2 deiodinase yields T 3 . Similarly, removal of the I from either the 3 or the 5 position of the inner ring of T 4 by type 3 deiodinase yields rT 3 . Further deiodination by any of the three enzymes ultimately yields T 0 (i.e., thyronine).
The relative activity of the outer-ring deiodinases changes in response to physiological and pathological stimuli. Caloric restriction or severe stress inhibits the type 1 deiodinase; this process decreases the conversion of T 4 to T 3 —and thus reduces the levels of T 3 . In contrast, levels of rT 3 rise by default in these situations, in part because of reduced conversion to DITs. These decreases in T 3 levels are accompanied by a decline in metabolic rate. You may think that because plasma levels of T 3 fall, there would be a compensatory rise in TSH, the secretion of which is inhibited by T 3 . However, because type 2 deiodinase mediates the conversion of T 4 to T 3 within the pituitary and CNS, and because caloric restriction does not affect this enzyme, local T 3 levels in the pituitary are normal. Thus, the thyrotrophs in the pituitary continue to have adequate amounts of T 3 , and no compensatory rise in TSH occurs. Teleologically, the rationale to restrain calorie expenditure in settings of decreased caloric intake is appealing. ![]() N49-2
N49-2
As noted in the text, calorie restriction inhibits type 1 monodeiodinase, reducing the conversion of T 4 to T 3 and thereby lowering circulating levels of T 3 . This effect of caloric restriction makes sense for someone who is starving because it tends to conserve body stores of fuel. On the other hand, this effect makes it more difficult to lose weight intentionally while dieting.
Thyroid hormones act on many body tissues to exert both metabolic and developmental effects. Once T 4 and T 3 leave the plasma, they enter the cell either by diffusing through the lipid of the cell membrane or by carrier-mediated transport ( Fig. 49-5 ). Most, but not all, of the actions of thyroid hormones occur as thyroid hormones bind to and activate nuclear receptors (see pp. 71–72 ). The multitude of thyroid hormone actions is mirrored by the ubiquitous expression of thyroid hormone receptors (TRs) throughout the body's tissues. There are actually two TR genes—α (chromosome 17) and β (chromosome 3)—and at least two isoforms of TRβ. The expression of these receptor genes is tissue specific and varies with time of development. The liver expresses TRβ, whereas TRα predominates in the brain. During development, the amount of α expressed may vary 10-fold or more. Both receptors bind to DNA response elements, predominately as heterodimers in association with the retinoid X receptor ( RXR ), and alter the transcription of specific genes.

Biologically, T 3 is much more important than T 4 . This statement may be surprising inasmuch as the total concentration of T 4 in the circulation is ~50-fold higher than that of total T 3 . Nevertheless, T 3 has greater biological activity for three reasons. First, T 4 is bound (only 0.01 to 0.02% is free ) more tightly to plasma proteins than is T 3 (0.50% is free ). The net effect is that the amounts of free T 4 and free T 3 in the circulation are comparable. Second, because the target cell converts some T 4 —once it has entered the cell—to T 3 , it turns out that T 4 and T 3 are present at similar concentrations in the cytoplasm of target cells. Third, the TR in the nucleus has ~10-fold greater affinity for T 3 than for T 4 , so that T 3 is more potent on a molar basis. As a result, T 3 is responsible for ~90% of the occupancy of TRs in the euthyroid state. ![]() N49-3
N49-3
Many hospitalized patients who are extremely ill exhibit abnormal results on thyroid function tests. However, the thyroid activity of most of these patients is actually appropriate and needs no correction. Many of these patients are in an intensive care unit (ICU) setting, and it is extremely important to distinguish true thyroid disease from this so-called sick euthyroid syndrome.
Sick euthyroid syndrome can take many forms, but the most common is a low or lower-than-normal total T 4 level and a low T 3 level. In true hypothyroidism, the diminished levels of thyroid hormones decrease feedback inhibition on the pituitary gland and lead to increased levels of TSH (see p. 1014 ). In sick euthyroid syndrome, the TSH level is usually normal. Although the reasons for this situation are not completely understood, at least one explanation may lie in the distinction between type 1 deiodinase, which is found in the periphery, and type 2 deiodinase, which is present in the pituitary. In sick euthyroid syndrome, the activity of type 1 or peripheral 5′/3′ deiodinase decreases, so that there is less conversion of peripheral T 4 to T 3 , but more conversion to rT 3 . As a result, peripheral T 3 levels fall. However, as was described in the main text with regard to starvation, type 2 deiodinase is not affected by stress nearly as much as is type 1 deiodinase; therefore, the pituitary gland continues to sense normal levels of T 3 , and it responds to what it perceives as normal levels of feedback inhibition from local T 3 on the production and release of TSH. However, other factors must also be involved, inasmuch as this mechanism does not adequately account for the decrease in total T 4 .
Patients with sick euthyroid syndrome may appear profoundly hypothyroid, exhibiting hypothermia and a sluggish sensorium, but they are not hypothyroid, and they should not receive thyroid hormone replacement. In fact, treating sick euthyroid patients with thyroid hormone yields either no improvement or a worse outcome.
When T 3 or T 4 binds to the TR in the nucleus, the hormone-bound receptor either activates or represses the transcription of specific genes. As discussed above, TR preferentially binds to DNA as a heterodimer of TR and RXR (see Table 3-6 ). TR belongs to the superfamily of nuclear receptors that may contain domains A through F (see Fig. 3-14 ). Three regions are especially important for TR: (1) The amino-terminal A/B region contains the first of two transactivation domains, which are responsible for transducing receptor binding into a conformational change in the DNA and thereby initiating transcription. (2) The middle or C region is responsible for DNA binding through two zinc fingers (see p. 82 ) as well as dimerization between receptors. (3) the E region, toward the carboxyl terminus, is responsible for binding the ligand (T 3 or T 4 ), and also for dimerization.
In addition to binding to receptors in the nucleus, T 4 and T 3 bind to sites in the cytosol, microsomes, and mitochondria. This observation has raised the issue of whether thyroid hormones exert actions through mechanisms not involving transcriptional regulation. Nongenomic actions of thyroid hormones have been observed in several tissues, including heart, muscle, fat, and pituitary. Thyroid hormones can act via nongenomic pathways to enhance mitochondrial oxidative phosphorylation—or at least energy expenditure as measured by O 2 consumption. Nongenomic targets of thyroid hormones include ion channels, second messengers, and protein kinases. It is less clear whether these actions occur via TRα or TRβ—similar to the nongenomic actions of estrogens, which involve the estradiol receptor (see p. 989 )—or whether other high-affinity thyroid-binding proteins are involved.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here