Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

Diffuse large B-cell lymphoma (DLBCL) is an aggressive lymphoma. In contrast to indolent (low-grade) lymphoma, the survival curve typically shows an initial downward slope followed by a plateau, indicating the potential curability of a significant proportion of patients who achieve remission ( Fig. 23-1 ).
In previous classifications, such as the Kiel classification and the working formulation, two major types of DLBCL were recognized: centroblastic (large non-cleaved cell) and immunoblastic lymphomas. In view of the low intraobserver and interobserver reproducibility in making the distinction, a single category of DLBCL was created to encompass both entities in the Revised European American Lymphoma (REAL) classification and the World Health Organization (WHO) classification. It was recognized, however, that DLBCL is a heterogeneous category from which clinically relevant entities might be delineated. Currently, many clinicopathologic variants, distinct subtypes, and distinct disease entities are recognized ( Box 23-1 ), although they account for only a minority of all DLBCLs. The remaining cases are referred to as DLBCL, not otherwise specified (DLBCL-NOS) in the 2016 WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues .
Morphologic variants
Centroblastic
Immunoblastic
Anaplastic
Other rare variants
Molecular subtypes
Germinal center B-cell-like (GCB)
Activated B-cell type
T-cell/histiocyte–rich large B-cell lymphoma
Primary DLBCL of the central nervous system (see Chapter 61 )
Primary cutaneous DLBCL, leg type (see Chapter 20 )
EBV + DLBCL, NOS (see Chapter 29 )
Large B-cell lymphoma with IRF4 rearrangement (provisional entity)
Primary mediastinal (thymic) large B-cell lymphoma
Intravascular large B-cell lymphoma
DLBCL associated with chronic inflammation (see Chapter 29 )
Lymphomatoid granulomatosis (see Chapter 29 )
ALK + large B-cell lymphoma (see Chapter 25 )
Plasmablastic lymphoma (see Chapter 25 )
HHV-8–positive diffuse large B-cell lymphoma (see Chapter 29 )
Primary effusion lymphoma (see Chapter 29 )
High-grade B-cell lymphoma, with MYC and BCL2 and/or BCL6 rearrangement
High-grade B-cell lymphoma, NOS (see Chapter 24 )
ALK, anaplastic lymphoma kinase; EBV, Epstein-Barr virus.
DLBCL is a diffuse proliferation of large or medium-sized neoplastic B cells with a nuclear size greater than or equal to that of a histiocyte nucleus, or more than twice the size of a small lymphocyte ( Fig. 23-2 ). Cases not conforming to the defined subtypes and entities are given the diagnostic label DLBCL-NOS .

DLBCL is the most common type of non-Hodgkin's lymphoma, accounting for 31% of all cases according to an international multicenter study. There is no significant difference in the incidence of this lymphoma among different ethnic and racial groups, except for certain specific subtypes of DLBCL (see later in the chapter). In some populations, such as Asians, DLBCL accounts for a higher percentage of all non-Hodgkin's lymphomas than in the United States and Western Europe (>40%), but this can be explained by a lower incidence of follicular lymphoma in these populations.
The median age of the patients is 64 years, but any age can be affected. There is a slight male predominance (male-to-female ratio of 1.2 : 1).
Most patients with DLBCL have no known underlying risk factors. A minority of cases occur in the setting of congenital immunodeficiency or acquired immunodeficiency, such as human immunodeficiency virus (HIV) infection, transplantation, methotrexate treatment for rheumatoid arthritis, and fludarabine treatment for low-grade B-cell lymphoma (see Chapter 55 ). These cases are commonly associated with Epstein-Barr virus (EBV). EBV-positive DLBCL can also supervene in angioimmunoblastic T-cell lymphoma as a result of the immune dysfunction accompanying the T-cell lymphoma. EBV-positive DLBCL, NOS (formerly EBV-positive DLBCL of the elderly), which occurs in patients without evidence of overt immunodeficiency, is believed to result from the subtle immunologic deterioration that occurs as part of the aging process (see Chapter 29 ).
Rare extranodal cases of DLBCL are associated with chronic inflammation or irritation, such as postmastectomy lymphedema, chronic suppurative inflammation in bone and skin, previous surgery and metallic implants, juxta-articular soft tissues in long-standing rheumatoid arthritis, and long-standing pyothorax. Many of these cases are associated with EBV with a type III latency and are considered a distinct entity (DLBCL associated with chronic inflammation) (see Chapter 29 ).
Most DLBCLs arise de novo, but some cases transform from an underlying low-grade lymphoma, such as follicular lymphoma, chronic lymphocytic leukemia–small lymphocytic lymphoma, lymphoplasmacytic lymphoma, marginal zone lymphoma, or nodular lymphocyte–predominant Hodgkin's lymphoma (NLPHL). Some cases of DLBCL occur synchronously or metachronously with classical Hodgkin's lymphoma.
Most patients present with rapidly enlarging lymph nodes or tumor masses in extranodal sites. About 30% of cases present in extranodal sites, and 71% have extranodal involvement during the course of the disease. Common primary extranodal sites include the gastrointestinal tract (especially the stomach) and Waldeyer's ring, but practically any organ can be involved, including the skin, central nervous system, mediastinum, and bone. Extranodal lymphomas of specific sites, especially of the skin and central nervous system, show distinctive clinical and biologic features (see Chapter 20, Chapter 60, Chapter 61 ).
Approximately half of the patients present with early-stage (stage I to II) disease, and one third have B symptoms. Bone marrow involvement occurs in 16%, and it may show concordant or discordant histology (see the section on prognostic factors later in the chapter for the clinical significance).
Involved lymph nodes or tissues show complete or partial effacement of architecture by diffuse infiltrates of lymphoma cells, often with coagulative necrosis and permeation into the surrounding tissues ( Figs. 23-3 and 23-4 ). Uncommonly, the lymphoma shows an interfollicular or sinusoidal pattern of nodal involvement (see Fig. 23-3, B ; Box 23-2 ). Exceptionally, the tumor cells form deceptively cohesive nodules, mimicking carcinoma (see Fig. 23-4, B ). There may be a “starry sky” appearance imparted by interspersed histiocytes with phagocytosed cell debris ( Fig. 23-5 ). Sclerosis may be present, especially in mediastinal and retroperitoneal tumors ( Fig. 23-6 ). The lymph nodes may show co-existing low-grade lymphoma, such as follicular lymphoma, chronic lymphocytic leukemia (CLL), or NLPHL ( Fig. 23-7 ).


Diffuse large B-cell lymphoma (DLBCL)
Sinusoidal DLBCL, CD30 +
Microvillous DLBCL
ALK + large B-cell lymphoma
DLBCL, NOS (uncommon)
Anaplastic large-cell lymphoma, ALK +
Anaplastic large-cell lymphoma, ALK −
Histiocytic neoplasms or tumor-like conditions
Langerhans cell histiocytosis
Rosai-Dorfman disease
Histiocytic sarcoma (uncommon)
Metastatic non-hematolymphoid malignancies (e.g., melanoma, carcinoma, germ-cell tumor)
ALK, anaplastic lymphoma kinase; DLBCL, diffuse large B-cell lymphoma; NOS, not otherwise specified.


.")
At extranodal sites, in addition to forming tumor masses, the lymphoma cells commonly infiltrate in an interstitial pattern, resulting in wide separation and loss of the normal specialized structures, such as gastric glands, salivary acini, seminiferous tubules, and thyroid follicles ( Fig. 23-8 ). Infiltration into the epithelium can occur, and mucosal ulceration is common. An underlying extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT) may be present.

Cytologically, DLBCL comprises large to medium-sized lymphoid cells with the morphologic features of centroblasts (large non-cleaved cells), immunoblasts, or cells with intermediate features. Centroblasts have round to oval vesicular nuclei, multiple membrane-bound small nucleoli, and a thin rim of amphophilic cytoplasm ( Fig. 23-9 ); they can show multilobated or angulated nuclei (see Fig. 23-9, B and C ). Immunoblasts have round or oval vesicular nuclei; a single large, centrally located nucleolus; and a broad rim of basophilic cytoplasm ( Fig. 23-10 ). The immunoblasts sometimes exhibit plasmacytoid features, with eccentrically located nuclei and paranuclear hof. Nonetheless, the lymphoma cells may not conform to these classic cell types, exhibiting hybrid features of centroblasts and immunoblasts, a huge cell size, a predominantly medium cell size, irregular nuclear foldings, elongated nuclei, voluminous cytoplasm in a cell with nuclear features of a centroblast, or clear cytoplasm ( Figs. 23-11 and 23-12 ).
 subtype.")



Cytologic subclassification of DLBCL is optional. Lymphomas with greater than 90% immunoblasts are considered the immunoblastic variant, whereas those with less than 90% immunoblasts are considered the centroblastic (large non-cleaved cell) variant ( Fig. 23-13 ). However, it can be difficult to decide whether a lymphoma cell is a centroblast or an immunoblast, and most DLBCLs contain a mixture of the two cell types or cells with intermediate features. The anaplastic variant comprises cells with bizarre pleomorphic nuclei, often with multinucleated forms, and abundant cytoplasm ( Fig. 23-14 ). It may also mimic metastatic carcinoma because of the cellular pleomorphism, cohesive growth, or sinusoidal infiltration. This variant behaves no differently than conventional DLBCL. Occasional cases of DLBCL show plasmacytic maturation, with lymphoma cells admixed with variable numbers of neoplastic mature-looking plasma cells ( Fig. 23-15 ).



In DLBCL, there can be variable numbers of reactive cells in the background, such as small lymphocytes (mostly T cells), plasma cells, histiocytes, and polymorphs. Cases with a prominent component of reactive T cells, usually with histiocytes, are categorized as T-cell/histiocyte–rich large B-cell lymphoma (see later). In rare cases, coalescing small clusters of epithelioid histiocytes are present, mimicking lymphoepithelioid T-cell lymphoma (Lennert's lymphoma) ( Fig. 23-16 ). Occasional cases may present initially with lymph node infarction ( Fig. 23-17 ).
 T-cell lymphoma.")

Some uncommon or histologically deceptive morphologic variants are listed in Table 23-1 ( Figs. 23-18 to 23-21 ). A summary of the clinical, morphologic, immunophenotypic, and genetic features of DLBCL is presented in Box 23-3 .
| Morphologic Variant | Main Pathologic Features | Tumors With Which It Might Be Confused |
|---|---|---|
| Myxoid stroma | Sheets, cords, or single lymphoma cells suspended in abundant myxoid stroma | Various types of myxoid sarcomas, such as extraskeletal myxoid chondrosarcoma, myxofibrosarcoma |
| Spindle cell morphology | Lymphoma cells have a spindly appearance due to spontaneous cellular spindling or molding by collagen; predilection for skin | Various types of spindle cell sarcomas, spindle cell carcinoma, desmoplastic melanoma |
| Signet ring cell morphology | Lymphoma cells have cytoplasmic vacuoles, which may be due to immunoglobulin accumulation or aberrant membrane recycling | Signet ring cell carcinoma, liposarcoma |
| Fibrillary matrix or rosette formation | Lymphoma cells associated with a prominent fibrillary matrix or rosette formation; because the fibrillary material is formed by interdigitating cytoplasmic processes (hence rich in cell membrane materials), it typically shows strong staining for leukocyte markers | Neural tumors, such as neuroblastoma, primitive neuroectodermal tumor |
| Abundant crystal-storing histiocytes | Lymphoma cells intermixed with histiocytes having ingested crystallized immunoglobulin | Rhabdomyoma |
| Marked tissue eosinophilia | Lymphoma cells intermixed with numerous eosinophils | Hodgkin's lymphoma, peripheral T-cell lymphoma |
| Microvillous DLBCL | Presence of numerous microvillous projections on ultrastructural examination; may show prominent sinusoidal growth pattern (see Box 23-2 ); CD20 + , CD30 − , EMA – , CD56 +/– | Anaplastic large cell lymphoma (CD20 − , CD3 +/– , CD30 + , EMA +/– , ALK +/– , CD56 –/+ ) |
| Sinusoidal CD30 + DLBCL | Sinusoidal growth pattern (see Box 23-2 ); CD20 + , CD30 + , EMA –/+ , ALK – | Anaplastic large-cell lymphoma (CD20 − , CD3 +/– , EMA +/– , ALK +/– ); microvillous DLBCL (CD30 − ); ALK + DLBCL (CD30 − , ALK + ); metastatic carcinoma (cytokeratin +); metastatic melanoma (S-100 + ) |




Median age: 64 years
Slight male predominance
Presents with rapidly growing nodal (70%) or extranodal (30%) tumor
B symptoms in one third of cases
Stage distribution: I, 25%; II, 29%; III, 13%; IV, 33%
Potentially curable: when treated by standard chemotherapy, complete remission can be achieved in two thirds of patients, with two thirds of them remaining relapse-free on long-term follow-up; the overall survival rate used to be 46%, but in recent years survival has improved by about 20% with the addition of rituximab to the chemotherapy regimen
Diffuse proliferation of large to medium-sized lymphoid cells, which can be indistinguishable from normal centroblasts or immunoblasts or can exhibit overt atypia such as irregular nuclear foldings, coarse chromatin, giant size, or bizarre nuclei
May be associated with an underlying low-grade lymphoma
Optional cytologic subclassification into centroblastic, immunoblastic, and anaplastic subtypes
Uncommonly, can show a variety of deceptive growth patterns (e.g., myxoid change, fibrillary matrix, spindle cells); see Table 23-1
Positive for pan–B-cell markers (e.g., CD20, CD22, CD79a, PAX5)
Positive for surface or cytoplasmic immunoglobulin
BCL6 + in ~60%
CD10 + in ~40%
CD5 + in ~5%-10%
CD30 + in ~15%
CD43 + in ~25%
BCL2 + in ~50%
Ki67 index: >20% (mean, 55%)
MYC + in ~40%
Clonally rearranged immunoglobulin genes
BCL2 rearranged in ~20%
BCL6 rearranged in ~30%
BCL6 mutated in ~70%
MYC rearranged in ~10%
Usually EBV – , except in the setting of immunodeficiency and the uncommon cases of EBV + DLBCL NOS
GCB type: Expresses genes characteristic of germinal-center B cells, and accounts for ~50% of all DLBCLs. More favorable prognosis than ABC type. Common genetic changes: BCL2 translocation, REL amplification, EZH2 mutation, mutations of genes in the Gα13 pathway.
ABC type: Expresses genes highly expressed during in vitro activation of peripheral blood B cells, and accounts for ~30% to 40% of all DLBCLs. Common genetic changes: NF-κB pathway activation through mutations in regulator genes of the NF-κB pathway, activation of B-cell receptor pathway, and MYD88 mutation.
Unclassified: Does not express genes characteristic of the GCB or ABC group at a high level
ABC, activated B cell; DLBCL, diffuse large B-cell lymphoma; EBV, Epstein-Barr virus; GCB, germinal-center B cells; NOS, not otherwise specified.
DLBCLs express CD45 and various pan–B-cell markers, such as CD20, CD22, CD79a, and PAX5. However, CD20 expression is lost in 60% of recurrent tumors from patients previously treated with rituximab (anti-CD20 chimeric antibody). Monotypic surface or cytoplasmic immunoglobulin (Ig) can frequently be demonstrated (IgM > IgG > IgA), and cytoplasmic immunoglobulin can sometimes be demonstrated as well. Pan–T-cell markers are negative, although CD3 is very rarely expressed ( Fig. 23-22 ).

CD10 expression occurs in 20% to 40% of cases. CD10 expression is useful in identifying the subset of DLBCL with a germinal-center B-cell expression profile, and this marker is often expressed in cases carrying the t(14;18) translocation. The reported positivity rate for the BCL6 protein is highly variable because the criteria for positive staining vary greatly from study to study. Overall, approximately 60% of cases are BCL6 positive, when only those with large aggregates of positive tumor cells are considered positive ( Fig. 23-22 , C ). Some cases of DLBCL express post–germinal-center or plasma cell–associated markers such as CD38, VS38, and IRF4/MUM-1, but CD138 expression is seen almost exclusively in cases showing morphologic evidence of plasmacytic differentiation, such as plasmablastic lymphomas (see Chapter 25 ).
The reported positivity rate for MYC protein expression in DLBCLs varies widely from 12% to 65% (~40% overall), attributable to the different cutoff values used (≥40% being the most popular), tumor heterogeneity, and whether very weakly stained cells are included in the count. MYC protein expression does not necessarily correlate with MYC rearrangement ; thus its possible role as a screening test to predict the presence of MYC rearrangement may be limited.
About 50% of cases express the BCL2 protein, and a higher frequency is observed in nodal than extranodal tumors. The variations in reported BCL2 positivity rate in DLBCL can be explained by the different cutoff values and the different antibodies used in different series. BCL2 expression in DLBCLs is associated with BCL2 translocation or amplification, but there are cases with a discrepancy between BCL2 translocation and BCL2 expression, which may be related to the type of antibody used and phosphorylation (rather than mutation) of BCL2 .
CD5 is expressed in approximately 5% to 10% of cases of DLBCL ( Box 23-4 ), and it is unclear whether de novo CD5-positive DLBCL represents a distinct clinicopathologic entity or merely an immunophenotypic variant of DLBCL, lacking relationship with CLL or mantle cell lymphoma and associated with adverse prognostic features. The median age of patients is in the seventh decade. De novo CD5-positive DLBCL can involve nodal and extranodal sites. It shows aggressive clinical features: a high proportion of patients have high IPI scores, more than 60% of patients have stage III or IV disease, and 75% have extranodal involvement (most frequently bone marrow). A proportion of patients have intravascular large B-cell lymphoma (see later) or show a splenic presentation. The morphology is indistinguishable from DLBCL-NOS. Most cases have a centroblastic appearance, and 19% have an intravascular or intrasinusoidal growth pattern. In the spleen, the infiltrate tends to be localized to the red pulp. Four morphologic variants—common-monomorphic, giant-cell–rich, polymorphic, and immunoblastic—have been described. They are usually CD10 negative. BCL2 and BCL6 are expressed in more than 80% of cases.
Mantle cell lymphoma, blastoid or pleomorphic variant
Paraimmunoblastic variant of chronic lymphocytic leukemia
DLBCL arising in chronic lymphocytic leukemia (Richter's syndrome)
Intravascular large B-cell lymphoma
Splenic DLBCL
De novo CD5 + DLBCL
The activation marker CD30 is expressed in 14% to 25% of cases, affecting some to most of the neoplastic cells. CD30 is potentially a target for therapy. CD43 is expressed in approximately 25% of DLBCLs. A small proportion of DLBCLs (~2% overall) express cyclin D1, often in only a proportion of tumor cells, with weak to moderate intensity. Such cases, with rare exceptions, do not express CD5 and SOX11 and lack the CCND1 gene translocation.
Ki67 staining in DLBCL usually shows a high proliferation index (>20%, but often <80%), and some cases may show an index approaching 100%.
DLBCLs have clonally rearranged immunoglobulin heavy-chain and light-chain genes (IGH, IGK, and IGL) and germline T-cell receptor (TCR) genes. The immunoglobulin heavy-chain variable region gene (IGHV) is usually hypermutated, with some cases showing ongoing somatic mutations.
The pathogenesis of DLBCL is complex because it involves at least two different pathways: a transformation pathway and a de novo pathway. Approximately 20% of cases of DLBCL show BCL2 rearrangement due to the t(14;18)(q32;q21) translocation, a hallmark of follicular lymphoma. Such cases may have transformed from a known or occult follicular lymphoma or may have evolved to DLBCL without a precursor phase of follicular lymphoma. BCL6 and other genes play an important role in the de novo pathway. BCL6 (3q27) rearrangement, which is found in some follicular lymphomas, occurs in about 30% of DLBCLs. The translocation partner can be the immunoglobulin genes, most commonly in the form of t(3;14)(q27;q32), or other genes. The BCL6 somatic mutation is a common event in DLBCL (73% of cases) and is unrelated to the presence or absence of BCL6 gene rearrangement. It is a marker for cells that have been through the germinal center and is thus commonly observed in various types of B-cell lymphomas corresponding to the germinal-center or post–germinal-center stage of differentiation, in contrast to earlier results suggesting that it was restricted to DLBCLs and follicular lymphomas. Persistent expression of the BCL6 protein as a result of BCL6 translocation or mutation inhibits differentiation and apoptosis, resulting in cellular proliferation.
MYC (8q24) rearrangement, a hallmark of Burkitt's lymphoma, occurs in about 10% of DLBCLs, being more common in HIV-infected patients, pediatric patients, and extranodal lymphomas. * It commonly occurs as part of complex genetic alterations, and the partner gene can be an IG gene or a non-IG gene. About 40% to 60% of cases with MYC rearrangement represent double/triple-hit lymphoma with co-existing BCL2 and/or BCL6 rearrangement, and such cases are reclassified as high-grade B-cell lymphoma, with BCL2 and/or BCL6 and MYC rearrangement (under the category of high grade B-cell lymphoma, NOS ) in the 2016 WHO classification. † (see Chapter 24 ) Double-hit lymphomas with MYC and BCL2 rearrangement are more common in germinal-center–type DLBCL. Other than translocation, increased copy number of MYC has been reported in 7% to 38% of cases.
* References .
† References .
Mutation of TP53 and immunoreactivity for p53 protein occur in 22% and 40% of DLBCLs, respectively, and there is no strict correlation between the two phenomena. The role of TP53 in the genesis of DLBCL is unknown, but it may be associated with histologic transformation from an underlying low-grade lymphoma in some cases.
The availability of next-generation sequencing technology in recent studies has helped unravel the genetic landscape of DLBCL. The coding genome contains on average 50 gene alterations per case, and mutations have been found in genes not previously implicated in DLBCL pathogenesis (see Fig. 23-23 ).
 pathogenesis.")
Association with EBV is uncommon in DLBCL (<10% in immunocompetent hosts), and this is seen more frequently in the anaplastic and plasmablastic variants or in elderly patients (see Chapters 25 and 29 ). There is a strong association with EBV in immunocompromised patients, however (see Chapter 55 ).
Using DNA microarrays to study gene-expression profiles, two groups of DLBCLs corresponding to different stages of B-cell differentiation (cell-of-origin [COO]) can be identified. One group expresses genes characteristic of germinal-center B cells—GCB type, and the other expresses genes normally induced during in vitro activation of peripheral blood B cells—activated B-cell (ABC) type. The GCB, ABC, and unclassifiable groups account for approximately 50%, 30% to 40%, and 15% to 20% of all DLBCLs, respectively. GCB type DLBCL shows frequent gains of 12q12, whereas ABC type DLBCL shows frequent trisomy 3, gains of 3q and 18q21-q22, and losses of 6q21-q22. The common genetic alterations occurring in the two types of DLBCL are summarized in Figure 23-23 . BCL2 translocation and REL amplification are detected almost exclusively in the GCB group, and mutations of EZH2 and genes of the Gα13 pathway are also common. In the ABC group, constitutive activation of the nuclear factor-κB (NF-κB) pathway is important in its pathogenesis. This can be mediated through various mechanisms: (1) mutations of various genes coding for regulators of the pathway (including positive regulators, such as TRAF3, TRAF5, and MAP3K7, and negative regulators, such as TNFAIP3, also known as A20 ) ; (2) activation of the B-cell–receptor signaling pathway, such as through CD79B, CARD11, and CD79A mutations ; and (3) MYD88 L265P mutation (a mutation commonly found in lymphoplasmacytic lymphoma, but is also present in ~30% of cases of ABC DLBCL). Genetic lesions preventing terminal determination (such as mutation, deletion, or transcriptional repression of PRDM1 ) are also common. MicroRNA-expression profiling also shows distinct signatures for GCB and ABC groups, and most cases unclassifiable by gene-expression profiling show a strong similarity to the ABC group by microRNA-expression profiling. Although immunoblastic and polymorphic centroblastic subtypes (centroblastic DLBCL with increased number of immunoblasts, but <90%) are found more often in the ABC group, they are also observed in the GCB group; the GCB and ABC groups are thus not strictly related to histologic subtypes.
The GCB group was reported to show a much better 5-year overall survival than the ABC group: 76% versus 16% in the original study, and 60% versus 35% in a larger follow-up series (treated with standard chemotherapy). The prognostic difference is maintained in the rituximab era (combination of rituximab and chemotherapy), with a 5-year overall survival rate of 80% versus 50%. Besides prognostic significance, determination of the cell of origin for DLBCL is also important for selecting target therapy. For example, there are ongoing clinical trials testing efficacy of drugs for the ABC type of DLBCL such as lenalidomide (an immunomodulatory drug), bortezomib (a proteasome that inhibits NF-κB) and Ibrutinib (inhibits Bruton tyrosine kinase in the B-cell receptor signaling cascade).
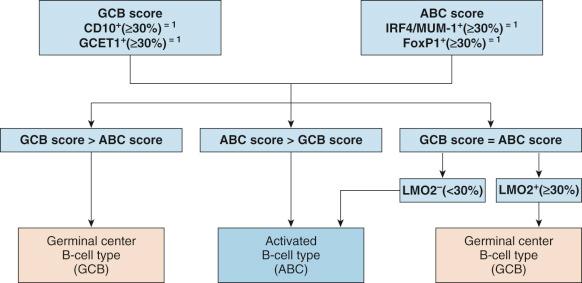
Because of issues of tissue requirement (fresh or frozen tissue), complexity, and low reproducibility of gene expression profiling using the microarray platform, various molecular methods adaptable to paraffin-embedded tissues have been developed, analyzing a limited panel of genes based on data from gene-expression studies to distinguish the GCB and ABC groups and establish signatures of prognostic significance. The most promising method appears to be the Lymph2Cx assay of twenty genes using the NanoString platform, which is robust and shows excellent interlaboratory agreement.
Various immunophenotyping algorithms have also been developed to determine the cell of origin for DLBCLs: GCB type versus ABC or non-GCB type ( Table 23-2 ). However, the correlation with gene-expression profiling results is imperfect, with concordance rate of 75% to 90%. There is also questionable interalgorithm concordance. Because the unclassifiable group cannot be recognized by immunophenotyping, such cases will be “forced” into either the GCB or ABC/non-GCB group. At least in some studies, DLBCLs classified by immunophenotypic algorithm into GCB and non-GCB types fail to show a prognostic difference for patients treated with a combination of rituximab and chemotherapy.
| Hans Algorithm | |||
| CD10 + (≥30%) | CD10 − (<30%) | ||
| BCL6 + (≥30%) IRF4/MUM-1 − (<30%) |
BCL6 + (≥30%) IRF4/MUM-1 + (≥30%) |
BCL6 − (<30%) | |
| Germinal-center B-cell type (GCB) | Non–germinal-center B-cell type (non-GCB) | ||
| Choi Algorithm | |||||
| GCET1 − (<80%) | GCET1 + (≥80%) | ||||
| CD10 + (≥30%) | CD10 − (<30%) | IRF4/MUM-1 + (≥80%) | IRF4/MUM-1 − (<80%) | ||
| BCL6 + (≥30%) | BCL6 − (<30%) | ||||
| FoxP1 − (<80%) | FoxP1 + (≥80%) | ||||
| Germinal-center B-cell type (GCB) | Activated B-cell type (ABC) | Germinal-center B-cell type (GCB) | |||
DLBCLs are derived from peripheral mature B cells at the germinal-center or post–germinal-center stage of differentiation. The GCB group shows ongoing somatic mutations in the IGHV gene, whereas the ABC group usually does not show ongoing mutations.
For de novo CD5-positive DLBCL, the presence of somatic hypermutation of IGHV, together with a low rate of ongoing somatic hypermutations and lack of CD10 expression, suggests post–germinal-center B-cell differentiation in about 80% of cases.
Although DLBCL is an aggressive tumor, usually resulting in death within 1 or 2 years if left untreated, it is a potentially curable disease. The survival curve tends to level off after 3 years, indicating curability of a substantial proportion of patients (see Fig. 23-1 ). DLBCL has historically been treated with combination chemotherapy, the standard being CHOP (cyclophosphamide, hydroxydaunomycin [doxorubicin], Oncovin [vincristine], and prednisone) or variants, with radiotherapy being added for bulky or localized tumors. Complete remission could be achieved in two thirds of patients, but one third of these successfully treated patients eventually relapsed. Those failing to achieve complete remission or achieving only partial remission died from the disease. The reported 5-year overall and failure-free survival rates were 46% and 41%, respectively. The addition of rituximab to the chemotherapy (R-CHOP) has improved overall survival by approximately 20%. There are ongoing studies to investigate the value of new drugs targeting specific deregulated pathways or molecules.
Pediatric patients with DLBCL have better outcome (3-year/5-year event-free survival of ~90%) compared with adult patients, and they used to be treated with aggressive chemotherapy regimens similar to those for patients with Burkitt's lymphoma. The difference in clinical behavior is at least partly related to the different biological features of DLBCL in children: more commonly of the GCB group (~80%), lack of BCL2 translocation, frequent MYC rearrangement (33%, associated with a more complex karyotype), and molecular signature of Burkitt's lymphoma in ~30% of cases. The use of strict age cutoff to determine treatment protocol has been challenged because this cannot be defined for the age-related biological characteristics. The role of rituximab in pediatric patients remains to be determined.
The adverse prognostic factors in DLBCL are listed in Box 23-5 .
High IPI score *
* Most important factors.
Immunoblastic or plasmablastic morphology
Lack of germinal-center cell phenotype (CD10 − , BCL6 − , LMO2 − , or following various cell-of-origin algorithms) *
Double expression of MYC and BCL2 *
CD5 expression
High proliferation (Ki67) index (controversial)
Lack of CD30 expression
CD43 expression
IRF4/MUM-1 expression
P53 expression
CD44s expression
P14 (ARF) nuclear overexpression
Cyclin D3 expression in ≥50% of lymphoma cells
Cyclin D2 expression
Protein kinase C-β expression
Lung-resistance protein expression
Survivin expression
Caspase 9 inhibition profile
Lack of HLA-DR expression
Poor tumor-infiltrating T-cell response, especially CD4 + or FOXP3 + T cells
High numbers of granzyme B + or TIA-1 + tumor-infiltrating T cells
Lack of SPARC + stromal cells (for ABC subgroup)
ALK expression (ALK + DLBCL; see Chapter 25 )
ABC type on gene-expression profiling *
Low HGAL expression
Low LMO2 expression
Redox signature score
BCL2 gene rearrangement
Lack of BCL6 gene rearrangement
Lack of BCL6 gene mutation
Low level of BCL6 messenger RNA transcripts
Non-IG/ BCL6 fusion
MYC gene rearrangement, especially IG/ MYC
Gain or increased copy number of MYC
TP53 mutation
Lack of hypermethylation of O 6 -methylguanine DNA methyltransferase promoter
Gains involving chromosome region 3p11-p12
ABC, activated B cell; ALK, anaplastic lymphoma kinase; IPI, International Prognostic Index.
The International Prognostic Index (IPI) is a reliable predictor of outcome ( Table 23-3 ). High IPI score is associated with poor outcome: the 5-year overall survival rate in high-risk patients is 22%, compared with 73% in low-risk patients. The revised IPI, based on redistribution of the individual IPI factors, has been reported to provide a better prediction of outcome in patients treated with R-CHOP.
| Prognostic Factors (1 Point Each) | |||||
| Age >60 yr Elevated serum lactate dehydrogenase Poor performance status High stage (III-IV) >1 Extranodal site |
|||||
| Risk Score | |||||
| 0 | 1 | 2 | 3 | 4 | 5 |
| Low | Low-intermediate | High-intermediate | High | ||
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here