Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

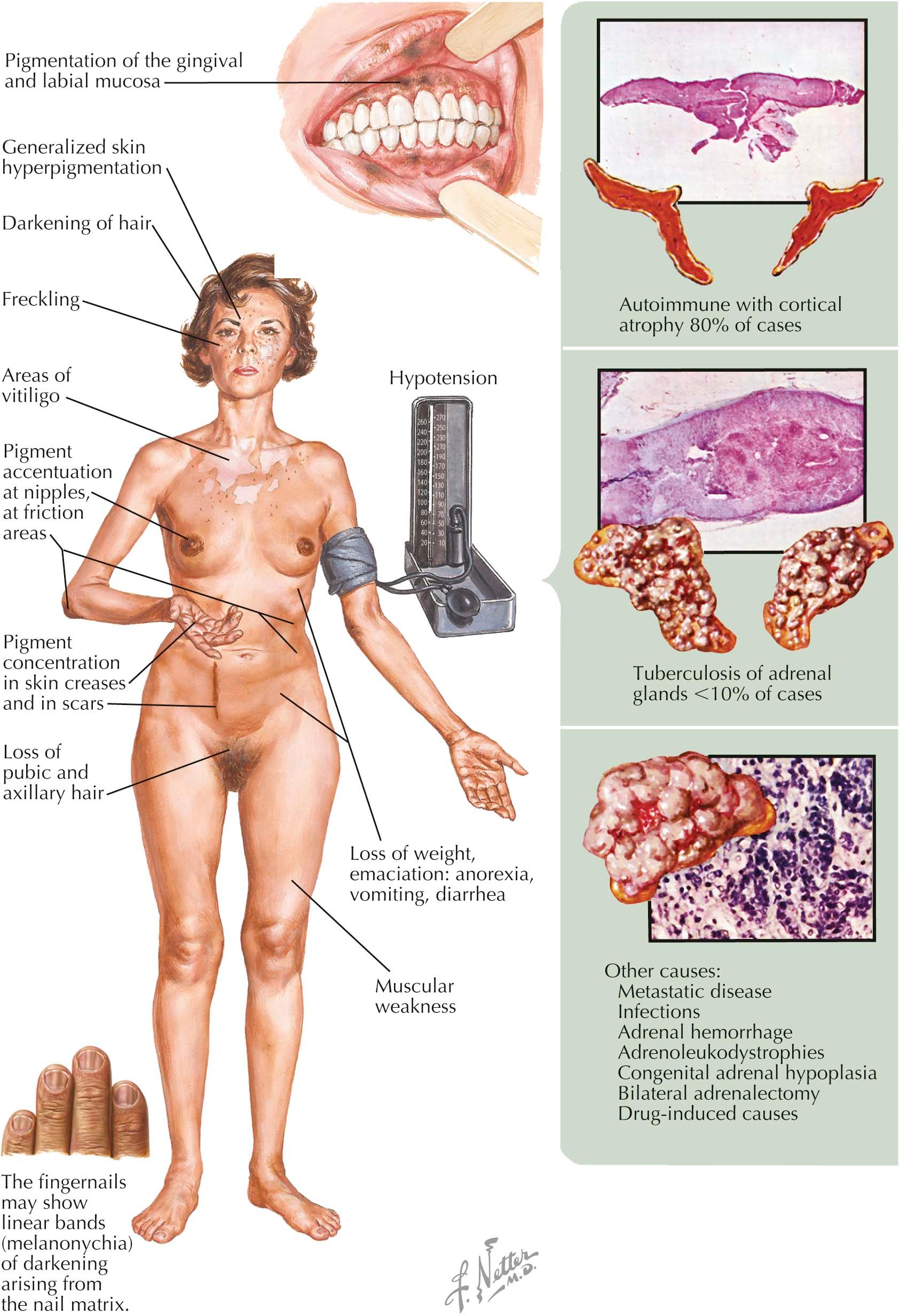
Addison's disease (chronic primary adrenocortical insufficiency) occurs when the adrenal gland has lost most of its functional capacity. Addison's disease can be caused by many different disease states that inhibit the functioning of the adrenal gland. The adrenal gland has a massive reserve capacity, and clinical manifestations of chronic adrenal insufficiency are not seen until the bilateral glands have lost at least 90% of their ability to produce adrenal hormones. Autoimmune destructive atrophy of the adrenal glands is the most common cause of Addison's disease. Infectious processes can cause destruction of the adrenal gland, with tuberculosis one of the more common causes of chronic adrenal gland insufficiency. Most cases of acute adrenal gland destruction are caused by bacteria (i.e., meningococcal disease).
Clinical Findings: Males and females are equally affected. The first symptoms are lethargy and generalized malaise. These symptoms may not be apparent until the affected patient undergoes a major stressful event, such as infection, which can lead to a prolonged disease course and a prolonged convalescence. Patients have excessive nervousness and may show emotional lability superimposed on periods of depression. Fatigue and weakness can be severe, to the point where even speaking causes fatigue. Weight loss and evidence of dehydration are present in most cases. Hypotension is frequently seen, and a small heart shadow is seen on chest radiography.
Cutaneous effects are always found in chronic primary adrenal insufficiency. Pigmentation is seen in many regions of the body and appears to occur in areas of friction, such as along the waist line and on elbows and knees. This is typically a generalized “bronze pigmentation,” but it is accentuated in the groin, nipples, and scrotum. The palmar and plantar creases are accentuated. Hyperpigmentation may be prominent within previous scars. Vitiligo may be present in conjunction with autoimmune adrenal insufficiency. Increased pigmentation of hair is seen, but this may be subtle and may occur slowly. Pigmentary alterations of the gingival and labial mucosa may also be seen. Pigmentary anomalies are not seen in secondary adrenal insufficiency, which is caused by pituitary deficiency.
Body hair is dramatically decreased, with near loss of axillary and pubic hair. The hair loss is more pronounced in females, because males still produce androgens, primarily in the unaffected testes. Serum testing shows hyperkalemia and hyponatremia, with a low cortisol level. The diagnosis is confirmed by intravenously injecting a synthetic corticotropin and evaluating the adrenal gland’s response by measuring cortisol levels after the injection. In patients with Addison's disease, the serum cortisol level is not increased by stimulation testing.
Histology: Skin biopsies are not helpful in making the diagnosis and are rarely performed. A normal number of melanocytes are present, with an increased amount of melanin pigment in the epidermis.
Pathogenesis: The adrenal glands are responsible for making cortisol, aldosterone, and the 17-ketosteroiods. When the adrenal glands no longer are able to produce these molecules, Addison's disease sets in. In the presence of low circulating levels of cortisol, the pituitary responds by increasing production of adrenocorticotrophic hormone (ACTH, corticotropin) and melanocyte-stimulating hormone (MSH). ACTH and MSH are derived from the same precursor protein, pro-opiomelanocortin (POMC). The pigmentary anomalies seen in Addison's disease are directly related to increased release of MSH. The increase in MSH causes pigment production by melanocytes in skin, hair, and mucous membranes. Pubic and axillary hair loss is related to the lack of 17-ketosteroids, whereas hypotension is caused by the lack of aldosterone. The lack of aldosterone causes a decreased blood volume and decreased serum sodium. The lack of cortisol production is responsible for weakness, fatigue, weight loss, and decreased mentation.
Addison's disease is seen frequently in association with other autoimmune endocrine disorders such as diabetes and autoimmune thyroiditis.
Treatment: Treatment requires the clearing of infection or treatment of the underlying cause of adrenal gland dysfunction. Supplemental hydrocortisone and fludrocortisone are used as replacement therapy for those with inadequate adrenal function. Hydrocortisone is used primarily to replace the missing cortisol, and fludrocortisone is used to replace aldosterone.
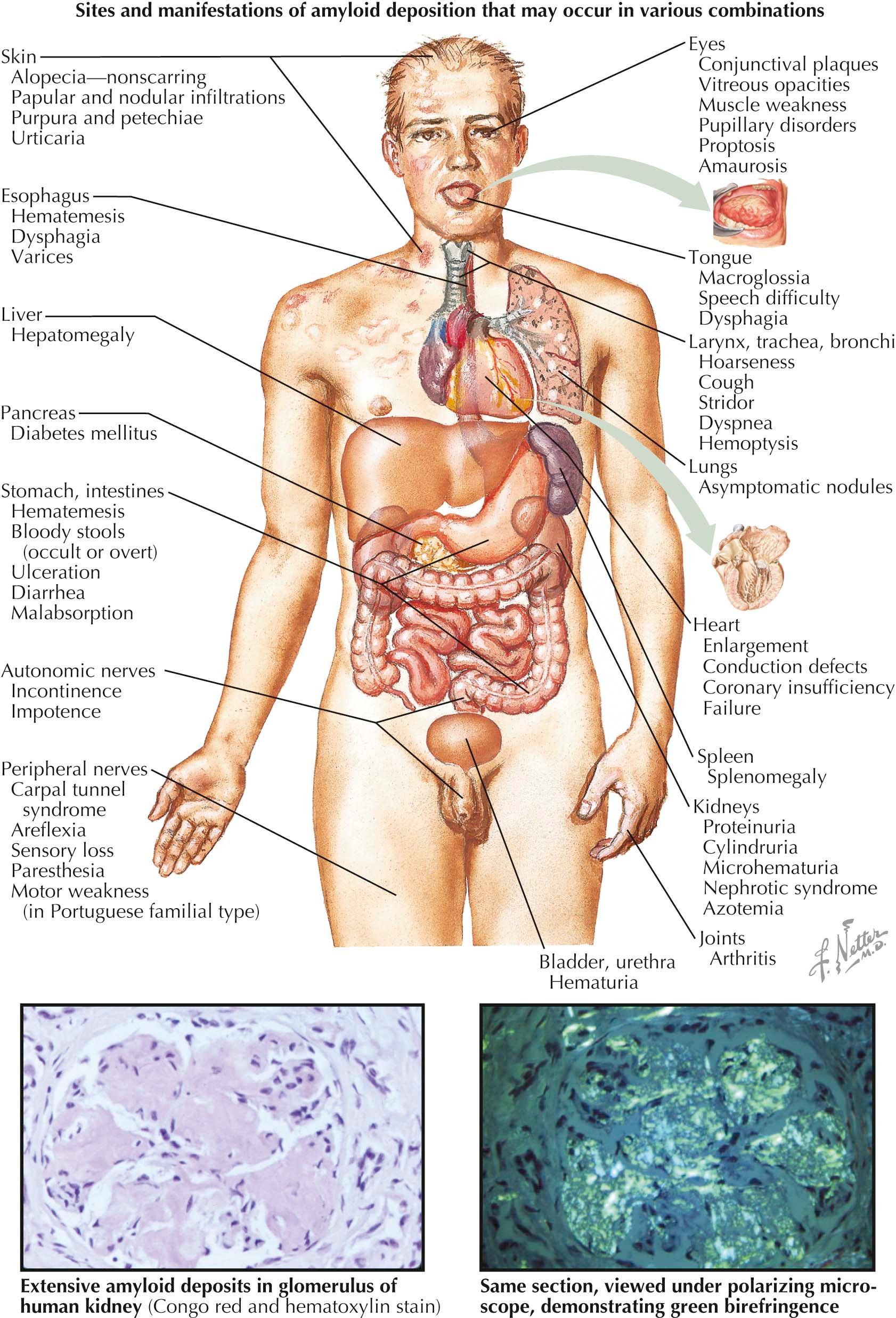
The term amyloidosis refers to a heterogeneous group of diseases. Systemic and cutaneous forms of amyloidosis can occur and are caused by the deposition of one of many different amyloid proteins. The primary cutaneous forms are more frequently seen. They include nodular, lichen, and macular amyloidosis (also referred to as lichen or macular amyloidosis). The systemic form is a multisystem, life-threatening disorder that requires systemic therapy. Most systemic disease is caused by an abnormality in plasma cells; myeloma-associated amyloid is a distant second in incidence. In addition to amyloidosis of the skin, the central nervous system may be involved with amyloidosis, as it is in Alzheimer's disease.
Clinical Findings: Systemic amyloidosis is caused by abnormal production of amyloid AL protein (immunoglobulin light chains) and its deposition in various organ systems. These effects can be seen in patients with plasma cell dyscrasia or myeloma. Mucocutaneous findings are often part of systemic amyloidosis, and on occasion they are the initial presentation of the disease. The hallmark cutaneous finding is translucent papules and plaques with varying degrees of hemorrhage. These papules are composed of the abnormal AL protein. Soft, rubbery papules may also occur within the oral mucous membranes. Pinch purpura of the skin is almost universal and results from weakening of the superficial cutaneous vessels by deposition of the AL protein. Periorbital ecchymoses may circumferentially surround the eye, which has led to the term “raccoon eyes.” The ecchymoses may be induced by coughing or by superficial trauma. The palms and soles may have a waxy appearance. The tongue is often strikingly enlarged due to amyloid deposits.
Deposition of the AL protein in close approximation to the dermal elastic fibers produces a rare finding termed amyloid elastosis. Clinically, this may mimic cutis laxa; the skin is easily distensible and lacks elastic recoil.
Deposition of amyloid in the renal glomeruli, liver, or heart muscle can cause significant end-organ damage. Renal insufficiency leading to renal failure is a major cause of morbidity and mortality. Hepatomegaly, leading to fibrosis and liver failure, may occur. Amyloid protein that is deposited in the muscle of the heart may lead to arrhythmias and congestive heart failure.
The primary cutaneous diseases known as lichen amyloidosis and macular amyloidosis are localized to the leg and the back, respectively. Most cases are believed to be directly caused by keratinocyte-derived amyloid protein. There are no systemic symptoms. Patients present with pruritic hyperpigmented macules and papules that may coalesce into plaques. Nodular primary cutaneous amyloidosis is caused by the local production of AL protein by plasma cells in the skin. This condition is extremely rare and may progress to systemic amyloidosis.
Pathogenesis: Systemic AL amyloidosis results from plasma cell dyscrasia or from myeloma-associated disease. It is directly caused by a proliferation of abnormal plasma cells. The plasma cells produce excessive amounts of immunoglobulin light chains, predominantly λ chains. The excessive amounts of AL protein are deposited within the walls of the cutaneous vasculature; this leads to weakening of the walls and is responsible for their easy rupture. The AL protein is deposited in many organ systems. Rarely, the plasma cells produce immunoglobulin heavy chains; this is termed AH protein.
Histology: Amyloidosis is a disease caused by the abnormal deposition of amorphous AL protein in the dermis and subcutaneous tissue. Biopsies of involved skin show eosinophilic deposits on routine staining. The amyloid protein is accentuated with special staining methods such as the Congo red stain. It shows an apple-green birefringence under polarized light microscopy and appears reddish under routine microscopy.
Treatment: Systemic amyloidosis is best treated with combination chemotherapy. Traditionally, prednisone and melphalan were the agents of choice. Newer proteosome inhibitors are currently used. Bone marrow transplantation is performed in certain cases.
Therapy for primary cutaneous amyloidosis is directed at symptomatic control. Topical corticosteroids and oral antihistamines are used to control itching. Varying results have been reported with ultraviolet phototherapy. No randomized, prospective studies of the treatment of primary cutaneous amyloid have been published.
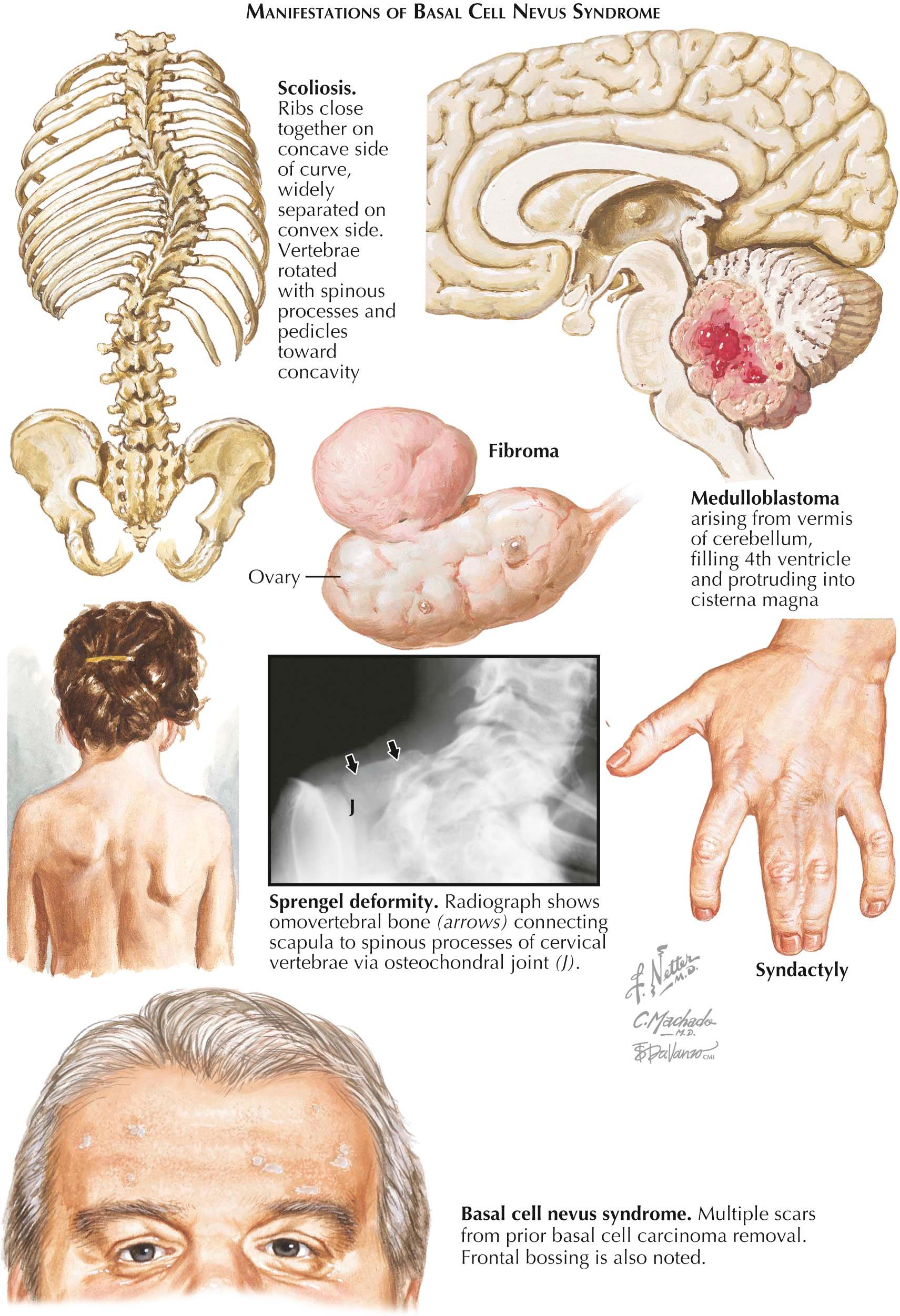
Basal cell nevus syndrome (BCNS), also known as nevoid basal cell carcinoma syndrome or Gorlin syndrome, is an uncommon autosomal dominant genodermatosis caused by mutations in the patched-1 (PTCH1) gene on chromosome 9. Approximately 40% of cases represent new, spontaneous mutations. Affected individuals are predisposed to the development of multiple basal cell carcinomas (BCCs), often in the hundreds over their lifetime. The diagnosis of this syndrome is based on a number of established criteria.
Clinical Findings: The incidence of BCNS is estimated to be 1 in 100,000 persons, and there is no race or sex predilection. It is inherited in an autosomal dominant fashion. Often, the first symptoms are painful keratogenic (odontogenic) jaw cysts. The early onset of BCCs often occurs before the age of 20 years.
Four of five BCNS patients have odontogenic jaw cysts on dental examination or dental radiographs. In children, the BCCs have been shown to mimic skin tags. Because skin tags are highly unusual in children, one should biopsy any skin tags seen in a young child to evaluate for BCC. About 90% of affected individuals show evidence of palmar pitting. This represents abnormal keratinization of the palmar skin. The lesions manifest as small (1-2 mm), pink to red, shallow defects in the glabrous skin of the palms or soles.
Medulloblastoma is uncommonly seen in patients with BCNS, occurring in only 1% to 2% of patients. Interestingly, 1% to 2% of children diagnosed with medulloblastoma are also diagnosed with BCNS. This is likely the most serious sequela of the syndrome and carries significant morbidity and mortality.
Diagnosis of BCNS is based on fulfillment of well-developed criteria. Two major criteria or one major and two minor criteria must be met to make the diagnosis. The six major criteria are (1) more than two BCCs; (2) palmar and plantar pitting; (3) odontogenic jaw cysts; (4) abnormalities of the ribs, including bifid or splayed ribs; (5) calcification of the falx cerebri; and (6) first-degree relative diagnosed with BCNS. The minor criteria are (1) congenital malformations (frontal bossing, hypertelorism, cleft palate, coloboma); (2) ovarian or cardiac fibromas; (3) macrocephaly; (4) skeletal abnormalities (scoliosis, syndactyly, Sprengel deformity of the scapula, pectus deformity); (5) medulloblastoma; and (6) other radiological abnormalities, including phalangeal lucencies in a flame shape and vertebral fusion.
Pathogenesis: BCNS is caused by to a defect in the PTCH1 gene on the long arm of chromosome 9. This gene is responsible for encoding the sonic hedgehog receptor protein that is found on many cell membranes. In normal physiological states, the transmembrane protein encoded by PTCH1 binds to the smoothened protein, turning off downstream cell signaling and ultimately decreasing cell proliferation. When the gene is mutated or when excessive sonic hedgehog protein is present, inhibition of the smoothened protein is removed, leading to uncontrolled cell signaling and a dramatically increased risk of cancer. Patients with BCNS are more sensitive to damage from ultraviolet light and radiation than normal controls.
Histology: BCC in the BCNS syndrome is histologically the same as any other BCC, and there are no distinguishing factors.
Treatment: BCCs tend to be multiple. Routine skin examinations and prompt removal of basal cell skin cancers help decrease the size of scarring and disfigurement resulting from surgery. All patients need to be educated at an early age on avoiding excessive sun exposure, tanning, and unnecessary radiation exposure from medical testing, because all of these increase the likelihood of BCC development. Many ongoing research protocols are looking at oral agents to decrease the abnormal hedgehog signaling pathway; such studies may lead to medical options for these patients in the future. Jaw cysts are best removed surgically to relieve pain and discomfort. Medulloblastoma is a serious, life-threatening tumor most commonly seen in early childhood, before the age of 4 years. Surgical and chemotherapeutic options exist.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here
