Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

Lymph Node and Organized Lymphoid Tissues, 1412
Diagnosis of Lymphomas, 1415
Hodgkin Lymphoma, 1428
Non-Hodgkin Lymphomas, 1444
Precursor Lymphoblastic Lymphoma, 1450
Mature B-Cell Lymphomas, 1453
Peripheral T-Cell and Putative NK-Cell Neoplasms, 1498
Lymphoproliferative Disorders Associated With Immunodeficiency, 1527
Tumors of Histiocytes and Dendritic Cells, 1529
Leukemia and Related Conditions, 1538
Metastatic Tumor in Lymph Node, 1539
Nonhematolymphoid Tumors and Tumorlike Lesions of Lymph Node, 1540
Practical Issues in Diagnosis of Lymphoproliferative Lesions, 1545
The lymph node is enclosed in a thin fibrous capsule, which is continuous with delicate fibrous trabeculae extending into the parenchyma. On the convex surface, afferent lymphatics drain into the subcapsular sinuses, which communicate through intermediate and medullary sinuses into efferent lymphatics that leave the node at the hilum. The sinuses are lined by sinus-lining cells and contain variable numbers of histiocytes and small lymphocytes. They are often distended with lymph in mesenteric nodes. The nodal parenchyma is supported by a delicate scaffolding formed by fibroblastic reticular cells, which may show variable immunoreactivity for cytokeratin and myoid markers such as desmin.
The cortex is the outer, convex portion of the node, in which B-cell lymphoid follicles are scattered ( Figs. 21A.1 and 21A.2 ). Primary follicles are round aggregates of small lymphocytes, which possess round or slightly irregular nuclei, condensed chromatin, and scanty cytoplasm. These lymphocytes are IgM+ and IgD+. The secondary follicle comprises a follicle center (germinal center) composed predominantly of centroblasts (large noncleaved cells) and centrocytes (small cleaved cells), which are IgD-, surrounded by a mantle of small lymphocytes (IgM+ and IgD+). The centroblasts possess round vesicular nuclei, multiple membrane-bound nucleoli, and a thin rim of amphophilic cytoplasm. The centrocytes have folded, angulated or triangular-shaped nuclei, fairly dense chromatin, indistinct nucleoli, and scanty cytoplasm (see Fig. 21A.1B ). These follicle center B cells (CD10+, BCL6+, HGAL+) are intermingled with tingible-body macrophages, follicular dendritic cells (FDC), and intrafollicular small T lymphocytes (predominantly follicular helper T cells, with a CD4+, CD10+, BCL6+, CXCL13+, PD1+, variably CD57+ phenotype). FDCs (CD21+, CD23+, CD35+) can be recognized by their thin violaceous nuclear membrane, empty nucleoplasm, small distinct nucleolus, and indistinct cell borders ; some may be binucleated. A marginal zone can sometimes be appreciated external to the mantle zone and is composed of midsize cells with a moderate amount of pale to clear cytoplasm (IgD-). The marginal zone is generally inconspicuous except in intraabdominal lymph nodes.
 Beneath the capsule and subcapsular sinus is the cortical zone containing lymphoid follicles. In the paracortical zone, venules are prominent. (B) The germinal center comprises a mixture of centroblasts (large noncleaved cells) and centrocytes (small cleaved cells). The latter cells have triangular-shaped or elongated nuclei, and barely visible cytoplasm.")
 Immunostaining for CD20 highlights nodular aggregates of B cells, which correspond to the follicles. The interfollicular zone contains only small numbers of B cells. (B) Immunostaining for CD3 highlights the T cells in the paracortical zone. Some T cells are also seen within the lymphoid follicles.")
In some reactive lymphadenopathies, monocytoid B-cells form a band or arc in the sinuses and around the reactive follicles. They are midsize cells with indented nuclei and abundant clear cytoplasm. There are commonly some admixed neutrophils.
The paracortex includes the portion of the node just deep to and between the follicles. It is populated mostly by T cells (small lymphocytes and larger blasts), which are mixed with some histiocytes, B lymphocytes (including immunoblasts), interdigitating dendritic cells, and Langerhans cells (see Fig. 21A.2 ). The latter two cell types can be recognized by the grooved or contorted nuclei and abundant lightly eosinophilic cytoplasm and are best highlighted by immunostaining for S100 protein. High endothelial venules lined by cuboidal endothelial cells are a characteristic feature of the paracortex; they represent the portal for lymphocyte trafficking.
The paracortex can harbor aggregates of plasmacytoid dendritic cells (CD68+, CD123+, CD303+, TCL1+), which are midsize, with eccentrically placed round nuclei, fairly dense chromatin, and an eccentric rim of amphophilic cytoplasm. There are typically interspersed apoptotic bodies and (sometimes) tingible-body macrophages.
The medulla represents the deep portion of the node, comprising plasma cell–rich medullary cords interspersed between the medullary sinuses. The medullary sinuses converge into the efferent lymphatics at the hilum, which is also the site where the arteries and veins drain into or out of the node.
The human body contains large amounts of nonencapsulated lymphoid tissues, which are mostly located in mucosal sites, such as the Waldeyer ring (ring of lymphoid tissue guarding the opening of the pharyngeal pathway), intestinal tract, and respiratory tract. They provide immunologic protection against infective agents and foreign antigens. These lymphoid aggregates have become known as the mucosa-associated lymphoid tissues (MALT). In contrast to lymph node, capsule and sinuses are typically absent. Architecturally there are two components: B-cell follicles separated by T-cell–rich interfollicular zones. The proportions of these two components and their degree of activity vary greatly from site to site and from time to time. There is generally a close interaction of the lymphocytes with the overlying epithelium.
There are three lineages of lymphocytes: B, T, and natural killer (NK). The stages of development of B and T lymphocytes and the changes in immunophenotype with cellular differentiation are shown in Figs. 21A.3 and 21A.4 . Many lymphoma types can be correlated with specific stages of lymphocyte maturation.
 are expressed in the cytoplasm (c-) or nucleus, while markers shown outside the circles are expressed on the cell surface (s-). Note that the common surface B–cell markers are lost with maturation into plasma cells.")
 are expressed in the cytoplasm (c-) or nucleus, while markers shown outside the circles are expressed on the cell surface.")
Lymphomas are neoplasms of the lymphoreticular system and can arise in lymph nodes or extranodal sites. They include (1) Hodgkin lymphoma (HL) and (2) non-Hodgkin lymphoma (NHL), with the latter being much more common.
To gain the maximum information from a tissue sample suspected of harboring lymphoma, proper handling of the tissue is essential: (1) making touch preparations for detailed cytologic assessment, (2) snap-freezing tissue for reserve in case frozen section immunohistochemistry or molecular analysis is required, (3) culture if necessary, and (4) sending tissue for other studies as appropriate (e.g., cytogenetics, flow cytometry). However, of utmost importance is leaving sufficient tissue for paraffin embedding for diagnostic purposes. Large lymph nodes must be thinly sliced to ensure adequate fixation. Buffered formalin is a good all-purpose fixative, affording good cytologic detail, good preservation of antigens for immunohistochemical detection, and fairly good preservation of messenger ribonucleic acid (mRNA) and deoxyribonucleic acid (DNA) for molecular studies. Although mercury-containing fixatives, such as B5, result in crisp nuclear details, their use has largely been phased out due to environmental concerns; while mercury-free formulations of these fixatives (e.g., B-Plus) exist, their use is nonetheless discouraged, as antigen preservation is unpredictable, and nucleic acid preservation is poor due to the presence of acid. Likewise, use of fixatives containing picric acid, such as Bouin solution, is discouraged because the resulting destruction of nucleic acids precludes molecular studies.
A good-quality hematoxylin and eosin (H&E)–stained section is essential for diagnostic purposes, although some pathologists prefer a Giemsa stain. A section of standard thickness (4 µm) is good for architectural assessment, and an additional thin section (2 µm) permits better assessment of cytologic details.
Since lymphoid tissue is fragile, it is highly prone to artifacts from compression or traction effects of biopsy, drying, overfixation, delayed fixation, etc. The outermost rim of the lymph node is often overfixed or subjected to drying, and thus the lymphoid cells may appear deceptively small and dark. The central portion of the node often suffers from delayed fixation, which results in blowing up of the nuclei and clearing of the chromatin. If such artifacts are present, the intermediate zone is the most optimal area for detailed cytologic assessment. Many mistakes in interpretation are committed due to suboptimal quality of the histologic sections obscuring the true pathologic process.
Malignant lymphoma can mimic or be mimicked by metastatic carcinoma or melanoma. On the other hand, it is most important not to mistake reactive lymphoid proliferations for lymphoma. The features helpful in making the distinction will be discussed under the various categories of lymphoma as well as at the end of this chapter.
Lymphomas, especially diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma, can undergo infarction spontaneously or after fine needle aspiration, rendering it difficult or impossible to render a diagnosis of lymphoma. The prior fine-needle aspiration materials, if available, should be reviewed to aid in diagnosis. If a diagnosis of lymphoma cannot be made, it is advisable to follow up and repeat biopsy of other nodes (if present) or nodes that subsequently appear, since total lymph node infarction may mask an underlying malignant lymphoma or metastatic malignancy.
Having made a diagnosis of lymphoma, the next step is to classify it. To categorize the cell size, the neoplastic cells can be compared with the reactive histiocytes interspersed among the lymphoma cells. Small, midsize, and large cells refer to cells with nuclei smaller than, approximately the same size as, and larger than those of the reactive histiocytes, respectively. If histiocytes are not found, the endothelial cell nuclei can be used instead as the “ruler,” although they are less satisfactory because of their ovoid or elongated shape. To classify a lymphoma, the cell size, nuclear configuration, cytoplasmic characteristics, cellular organization (such as follicular vs diffuse), and other subsidiary features (including clinical, laboratory, immunophenotypic, and/or molecular findings) must be considered.
Immunohistochemical studies are essential for the diagnosis of lymphoma. In particular, they are required for distinction from reactive lymphoid hyperplasia (if relevant), lineage determination, classification, and prognostication.
Immunostaining on frozen tissue has the advantage that practically all antibodies work on such tissues, but the main drawbacks are the additional handling procedures and the occasional difficulties in correlating staining results with cellular components. Nowadays, almost all leukocyte antigens can be reliably and consistently demonstrated on paraffin sections as a result of advances in antigen retrieval techniques and the availability of an expanding range of antibodies. Because of the convenience in handling and excellent cytomorphologic preservation, paraffin section immunohistochemistry is currently the preferred routine technique. Flow cytometry is an alternative technique for immunophenotyping and has largely replaced frozen section immunohistochemistry as the preferred means to assess expression of markers not amenable to immunohistochemistry on paraffin sections (such as surface CD3). Fresh tissue is required, and cells have to be disaggregated to produce a cell suspension, which is then incubated with fluorochrome-labeled antibodies before being analyzed by the flow cytometer. A scattergram is produced, indicating the size and fluorescence intensity of each individual cell. Gating is performed to analyze specific cell populations. The advantages of flow cytometric immunophenotyping include (1) prompt availability of results; (2) ability to analyze multiple markers simultaneously, facilitating identification of cell populations with special or aberrant immunophenotype; and (3) superior performance in detecting surface immunoglobulin (Ig) expression because the cells have been washed free from the interstitial serum fluids—contrasting with the frequent difficulties in interpretation of Ig staining in frozen or paraffin sections due to heavy interstitial staining. The main disadvantages are (1) requirement of fresh tissue, (2) lack of direct correlation with topography or cytology (e.g., a thymoma can potentially be mistaken for T–lymphoblastic lymphoma because the tumor sample is rich in T cells with an immature phenotype), and (3) underrepresentation of the neoplastic population in the cell suspension due to sclerosis or low tumor cell viability. Thus when the results contradict the histologic features, findings from other investigations have to be given more weight .
The antibodies that are useful for the evaluation of hematolymphoid proliferations and subtyping of lymphomas are listed in Tables 21A.1 and 21A.2 . The cluster of differentiation (CD) terminology is used where available ; antibodies that recognize the same antigen (protein molecule) have been assigned specific CD numbers at International Leukocyte Typing Workshops.
| CD (Antibodies) | Pattern of Staining | Main Reactivities in Normal Hematolymphoid Cells | Main Reactivities in Hematolymphoid Neoplasms | Remarks or Caution in Interpretation of Staining |
|---|---|---|---|---|
| GENERAL LEUKOCYTE MARKER | ||||
| CD45RB/leukocyte common antigen | Cell membrane, sometimes Golgi |
|
|
|
| B–LINEAGE ASSOCIATED | ||||
| CD19 | Cell membrane |
|
|
|
| CD20 (e.g., L26) | Cell membrane |
|
|
|
| CD22 | Golgi and/or cell membrane |
|
|
|
| CD23 | Cell membrane |
|
|
|
| CD79a | Diffuse cytoplasmic, often with accentuation around nuclear membrane |
|
|
|
| CD200 | Cell membrane |
|
|
|
| LEF1 | Nuclear |
|
|
|
| PAX5 (B-cell specific activator protein) | Nuclear |
|
|
|
| OCT2 | Nuclear |
|
|
|
| BOB.1 | Nuclear +/- cytoplasmic |
|
|
|
| Immunoglobulin (IgM, IgG, IgA, IgD, κ, λ) | Cytoplasmic with Golgi and perinuclear accentuation; or cell membrane |
|
|
|
| T–LINEAGE ASSOCIATED | ||||
| CD2 | Cell membrane +/- Golgi |
|
|
|
| CD3, surface (currently demonstrable only by flow cytometry or frozen section immunohistochemistry) | Cell membrane |
|
|
|
| CD3ε (cytoplasmic CD3) | Cytoplasmic with perinuclear or Golgi accentuation; rarely cell membrane |
|
|
|
| CD4 | Cell membrane |
|
|
|
| CD5 | Cell membrane |
|
|
|
| CD7 | Cell membrane |
|
|
|
| CD8 | Cell membrane |
|
|
|
| CD43 | Cell membrane; rarely Golgi |
|
|
|
| CD45RO | Cell membrane; rarely Golgi |
|
|
|
| LAT | Cell membrane |
|
|
|
| αβTCR (e.g., βF1) | Cell membrane |
|
|
|
| γδTCR (e.g., TCRCγM1, human pan TCRγδ1) | Cell membrane |
|
|
|
| NK-CELL ASSOCIATED | ||||
| CD56 | Cell membrane |
|
|
|
| CD57 | Cell membrane |
|
|
|
| CYTOTOXIC MARKERS | ||||
| TIA1 | Cytoplasmic granular |
|
|
|
| Granzyme B | Cytoplasmic granular |
|
|
|
| Perforin | Cytoplasmic granular |
|
|
|
| ACTIVATION AND PROLIFERATION ASSOCIATED | ||||
| CD25/interleukin 2 receptor | Cell membrane |
|
|
|
| CD30 | Cell membrane and/or Golgi |
|
|
|
| Ki67 | Nuclear |
|
|
|
| DIFFERENTIATION STAGE ASSOCIATED | ||||
| TdT/terminal deoxynucleotidyl transferase | Nuclear |
|
|
|
| CD10 | Cell membrane |
|
|
|
| CD99a/MIC2 | Cell membrane |
|
|
|
| BCL2 | Perinuclear space |
|
|
|
| BCL6 | Nuclear |
|
|
|
| HGAL | Cytoplasm |
|
|
|
| CD103/mucosal lymphocyte antigen | Cell membrane |
|
|
|
| EMA/epithelial membrane antigen | Cell membrane and/or Golgi |
|
|
|
| CD138/syndecan–1 | Cell membrane |
|
|
|
| MUM1 | Nuclear and cytoplasmic |
|
|
|
| FOLLICULAR T-HELPER CELL | ||||
| PD1 (CD279) | Cell membrane |
|
|
|
| CXCL13 | Cytoplasm, sometimes with punctate quality |
|
|
|
| LYMPHOMA ASSOCIATED | ||||
| Cyclin D1 | Nuclear |
|
|
|
| SOX11 | Nuclear |
|
|
|
| ALK/anaplastic lymphoma kinase | Nuclear or cytoplasmic |
|
|
|
| MYC | Nuclear |
|
|
|
| HISTIOCYTE, DENDRITIC CELL, AND MYELOID CELL ASSOCIATED | ||||
| CD68 | Cytoplasmic granular |
|
|
|
| CD163 | Cell membrane +/- cytoplasmic |
|
|
|
| S100 protein | Nuclear, with or without additional diffuse cytoplasmic staining |
|
|
|
| CD1a | Cell membrane |
|
|
|
| Langerin (CD207) | Cell membrane + cytoplasmic (granular) |
|
|
|
| CD15 | Golgi and/or cell membrane |
|
|
|
| CD21/C3d receptor | Cell membrane |
|
|
|
| CD35/C3b receptor | Cell membrane |
|
|
|
| Clusterin | Cell membrane |
|
|
|
| CD123 (interleukin-3 receptor, low affinity) | Cell membrane |
|
|
|
| Lysozyme | Cytoplasmic granular |
|
|
|
| Myeloperoxidase | Cytoplasmic granular |
|
|
|
| CD33 | Cell membrane |
|
|
|
| Problem to Be Addressed | Recommended First-Line Antibodies |
|---|---|
| B lineage? | CD20 (or PAX5, CD19) |
| T lineage? | CD3 (or CD2) |
| NK lineage? | CD56 (positive), surface CD3 (negative), cytoplasmic CD3 (positive), TCR (negative) |
| Myeloid cell? | Myeloperoxidase |
| Follicular center cell? | CD10 (or BCL6, HGAL, STMN1) |
| Follicular lymphoma or follicular hyperplasia? | BCL2, CD10 (interfollicular invasion) |
| Chronic lymphocytic leukemia? | CD5, CD23, LEF1 |
| Normal mantle zone cells? | IgD |
| Mantle cell lymphoma? | Cyclin D1 (+/- SOX11), CD5 |
| Burkitt lymphoma? | Ki67, CD10 (+/- BCL6), BCL2 (negative), MYC |
| Immature (precursor lymphoblastic) cell? | TdT |
| Anaplastic large cell lymphoma? | CD30, ALK |
| Plasma cell? | CD20 (negative), CD138 (positive), MUM1 (positive) |
| Histiocyte? | CD68 (or CD163) |
| Interdigitating dendritic or Langerhans cell? | S100 (also langerin/CD207 for the latter cell type) |
| Follicular dendritic cell? | CD21 (or CD23, CD35) |
| Hodgkin lymphoma? | CD30, CD15, PAX5 |
Immunohistochemical staining must be interpreted in the context of the morphologic findings and carefully correlated with the cell populations. For many lymphoma types, there can be many reactive cells in the background, and one must determine which markers are truly expressed in the atypical (neoplastic) cells. The localization of the signal in the cell must be appropriate for the antibody being used, otherwise the result cannot be considered positive (e.g., nucleolar staining for CD20 or diffuse cytoplasmic staining for CD30 is not considered true positive staining) ( Fig. 21A.5 ). When staining is negative, false negative staining must be excluded by ascertaining the presence of appropriate internal and/or external controls. In challenging cases, multiplex immunostaining may help in determining cell type–specific expression (e.g., costaining for CD5 and PAX5 to distinguish CD5 expression on B cells in chronic lymphocytic leukemia/small lymphocytic lymphoma from normal CD5 expression on T cells).
 with or without diffuse cytoplasmic staining is always significant, as it indicates cellular immunoglobulin synthesis. Staining in the perinuclear space is also indicative of cellular immunoglobulin synthesis (not shown).")
Uncommonly, CD3 positivity in B-cell lymphoma or CD20 positivity in T-cell lymphoma can occur, which may result in mistakes in lineage assignment for lymphomas. Some of the CD3+ B–cell lymphomas do not express conventional B-lineage markers (such as CD20 and PAX5) due to the plasmacytic/plasmablastic nature of the neoplastic cells, further compounding the problem. Application of additional B and T cell markers is needed to clarify the cellular lineage. For lymphomas arising in the setting of immunodeficiency, simultaneous expression of B and T lineage markers sometimes occurs, and it can be difficult to determine if such lymphomas are genuinely bilineage or merely B or T lymphomas with aberrant antigen expression.
Specific chromosomal translocations have been identified in some lymphoma types. They have been instrumental in shaping the classification of lymphomas, and their detection (by cytogenetics or other molecular techniques) can aid in diagnosis ( Table 21A.3 ). These translocations often, but not always, involve juxtaposition of a protooncogene with the immunoglobulin gene (heavy chain gene on chromosome 14, or light chain genes on chromosomes 2 and 22), resulting in overexpression of a structurally normal protooncogene product. Other chromosomal translocations result in the production of chimeric proteins that are constitutively activated.
| Lymphoma Type | Specific Chromosomal Translocation | Oncogene or Tumor Suppressor Gene Being Implicated | Mechanism of Lymphomagenesis |
|---|---|---|---|
| Follicular lymphoma | t(14; 18)(q32; q21) | BCL2 | Overexpression of BCL2 as a result of juxtaposition of BCL2 gene with IGH |
| Mantle cell lymphoma | t(11; 14)(q13; q32) | CCND1 | Overexpression of cyclin D1 as a result of juxtaposition of CCND1 gene with IGH |
| Extranodal marginal zone lymphoma of MALT | t(11; 18)(q21; q21) | BIRC3 , MALT1 | Production of chimeric protein BIRC3-MALT1, which activates NFκB |
| t(1; 14)(p22; q32) | BCL10 | Overexpression of BCL10 as a result of juxtaposition of BCL10 gene with IGH | |
| t(14; 18)(q32; q21) | MALT1 | Overexpression of MALT1 as a result of juxtaposition of MALT1 gene with IGH | |
| t(3; 14)(p14.1; q32) | FOXP1 | Overexpression of FOXP1 as a result of juxtaposition of FOXP1 gene with IGH | |
| Burkitt lymphoma | t(8; 14)(q24; q32) | MYC | Deregulated expression of MYC due to juxtaposition of MYC gene with IGH, IGK, or IGL |
| t(8; 22)(q24; q11) | |||
| t(2; 8)(p12; q24) | |||
| Diffuse large B-cell lymphoma (DLBCL) | Translocation involving 3q27 with a number of chromosome partners | BCL6 | Overexpression of BCL6 as a result of juxtaposition of BCL6 gene with IGH or other genes |
| t(14; 18)(q32; q21) | BCL2 | Overexpression of BCL2 as a result of juxtaposition of BCL2 gene with IGH | |
| Translocation involving 8q24 (occurring in an aggressive subset of DLBCL) | MYC | MYC fused with IGH or other genes, resulting in overexpression of MYC | |
| ALK+ large B-cell lymphoma | t(2; 17)(p23; q23) and other translocations involving 2p23 | ALK | Overexpression of a chimeric protein CLTC-ALK with constitutive tyrosine kinase activity |
| Large B-cell lymphoma with IRF4 rearrangement | Cryptic rearrangement of IRF4 with an IGH locus | IRF4 | Overexpression of MUM1 (IRF4) |
| T–lymphoblastic lymphoma/leukemia | t(1; 14)(p32-34; q11) | TAL1 | Overexpression of TAL1 as a result of juxtaposition of TAL1 gene with TR gene |
| ALK+ anaplastic large cell lymphoma | t(2; 5)(p23; q35) | NPM, ALK | Production of a chimeric protein NPM-ALK with constitutive tyrosine kinase activity |
| Variant translocations involving 2p23, e.g., t(1; 2), t(2; 3), inv(2)(p23; q35) | ALK and other partner genes | Production of a chimeric ALK protein with constitutive tyrosine kinase activity | |
| ALK- anaplastic large cell lymphoma | t(6; 7)(p25.3; q32.3) | DUSP22 | — |
| Inv(3)(q26q28) | TP63, TBL1XR1 | — | |
| Primary cutaneous anaplastic large cell lymphoma | Translocation involving 6p25.3 | DUSP22-IRF4 locus | — |
| Peripheral T-cell lymphoma, NOS | t(5; 9)(q33; q22) (predominantly in follicular variant) | ITK, SYK | Production of a chimeric kinase ITK-SYK kinase mimicking T-cell receptor signal |
IG (immunoglobulin) and TR (T cell receptor) gene rearrangements are the hallmarks of B cells and T cells, respectively, and are therefore sensitive and specific indicators of the lineage of a lymphoid proliferation. The native IG gene is formed by many segments of variable, diversity, and joining regions in addition to the constant region (germline configuration). When a cell is committed to B lineage, the IG gene is rearranged so that one of the many variable regions combines with one of the many diversity regions, one of the many joining regions, and a constant region, in a pattern specific for that cell; this is to create diversity in the Ig being produced. In B-cell lymphoma, which is a clonal proliferation, all the neoplastic cells carry an identical IG gene rearrangement. The principles are the same for TR gene rearrangement in T cells, because the TR gene is similarly composed of variable, diversity, and joining regions.
In routine practice, molecular studies are usually performed only when morphologic and immunohistochemical studies yield inconclusive findings. They can provide the following information :
Clonality of the lymphoid proliferation. Molecular studies can help in distinction of lymphoma from reactive lymphoid hyperplasia.
Lineage of the lymphoid proliferation. Practically all B-cell lymphomas show rearrangements of both IG heavy chain ( IGH ) and IG light chain ( IGH, IGK, IGL ) genes, while most T-cell lymphomas show rearrangements of TR β-chain ( TRB ) and γ–chain ( TRG ) genes. NK cell lymphomas typically lack rearrangements of both IG and TR genes. Exceptions do occur, and these are discussed under the individual tumor types.
Chromosomal translocations (gene fusions) characteristic of certain lymphoma types (e.g., BCL2 rearrangement in follicular lymphoma).
Gene mutations characteristic of specific hematolymphoid neoplasms (e.g., TET2 and IDH2 mutation in angioimmunoblastic T-cell lymphoma, and BRAF mutation in hairy cell leukemia).
Clonal relationship of different lymphoma components found concurrently or at different time points, either by comparing the molecular size of the rearranged IG or TR gene or, more accurately, by comparing the DNA sequences of the rearranged IG or TR gene.
Molecular alterations with prognostic or therapeutic implications for specific lymphoma types (e.g., MYC rearrangement as an adverse prognostic factor in DLBCL).
Detection of minimal residual disease.
Southern blot analysis has been the gold standard method for detection of IG or TR rearrangements and chromosomal translocations in lymphomas, but it has largely been replaced by other techniques (such as polymerase chain reaction [PCR]) because it is demanding in terms of time and specimen requirement (fresh or frozen tissue).
PCR offers a highly sensitive and versatile molecular technique that is widely used in the diagnostic laboratory. This is applicable on routine paraffin-embedded tissues, even when only minute quantities of tissue are available.
To detect IG or TR gene rearrangements, primers recognizing the conserved sequences of these genes are used for PCR. After gel electrophoresis of the amplicon, the DNA product can be stained with ethidium bromide and visualized under ultraviolet light. If a clonal lymphoid population is present, a sharp band (or two sharp bands derived from the two alleles of the IG or TR gene) of the expected molecular size can be visualized because the amplified DNA products are of identical molecular size ( Fig. 21A.6 ). If the population is polyclonal, a ladder (many bands) or smear pattern is observed because the amplified DNA products from the different cells are of different molecular sizes and thus migrate to different positions during electrophoresis (see Fig. 21A.6 ). Neoplastic clonal populations comprising as low as 1% of the specimen can be detected. On the other hand, there is a significant false negative rate of 10% to 40% compared with Southern blot analysis. The false negative result is attributable to failure of the so-called universal primers to hybridize with the rearranged IG or TR gene due to partial rearrangements or somatic hypermutations. The false negative rate is higher for germinal center and post–germinal center B-cell neoplasms (such as follicular lymphoma and extranodal marginal zone lymphoma) because the rearranged IG genes show somatic hypermutations. Nonetheless, the false negative rate can be lowered significantly by using multiple primer pairs targeting the different regions of the rearranged genes, such as the BIOMED-2 multiplex PCR strategy. In recent years, analysis of the PCR products by capillary electrophoresis and GeneScan has become more popular because the product size and quantity can be determined more accurately. For monoclonal lesions, one or two sharp peaks are observed, while for polyclonal (reactive) lesions, gaussian curves with triplet spacing of peaks are observed.
 for immunoglobulin gene. After PCR, the gel is stained with ethidium bromide and visualized under ultraviolet light after gel electrophoresis of the amplified DNA. Lane M: molecular size markers. Lane 1: multiple bands seen (ladder pattern), consistent with a polyclonal pattern. Lanes 2 to 4: a strong discrete band seen in a background of many weaker bands, indicating presence of a single B-cell clone in a reactive B-cell background; this supports a diagnosis of B–cell lymphoma. Lanes 6 and 7: positive control from a known example of B–cell lymphoma; the single sharp band indicates presence of clonal immunoglobulin gene rearrangement.")
PCR can be used to detect chromosomal translocations through the use of primers specific for the genes involved in the translocation (such as BCL2 and IGH genes in follicular lymphoma); successful amplification indicates the presence of the gene fusion, while lack of amplicons indicates absence of gene fusion. However, this is not the method of choice for demonstration of chromosomal translocations in lymphomas because of the relatively high false negative rates due to marked variabilities in the breakpoints and a potential false positive result due to the presence of low-level gene fusion positive cells (such as BCL2-IGH and NPM-ALK ) in some normal subjects. Fluorescence in situ hybridization (FISH) is the preferred method (see next section).
Gene mutations (such as IDH2, TET2, BRAF, MYD88 ) can be detected in tumor samples by PCR direct sequencing or allele-specific PCR, or by use of targeted next-generation sequencing panels.
Interphase FISH, which is applicable on fresh, frozen, or paraffin-embedded tissue, is the technique of choice for detection of specific chromosomal translocations in lymphomas because of its high sensitivity and specificity. Depending on the choice of probes, one looks for comigration of two signals (indicating fusion of genes) or separation of the signals (indicating breakpoint of a genetic locus) to indicate the presence of chromosomal translocation. FISH is also a convenient method to detect monosomy or trisomy of chromosomes, gene deletion, or gene amplification.
Chromogenic in situ hybridization (CISH) permits visualization of the signals by light microscopy. The most commonly used probes for the diagnosis of hematolymphoid lesions include Epstein-Barr virus early RNA (EBER) and immunoglobulin light chains (κ and λ). In situ hybridization for EBER is the most sensitive and specific technique for demonstration of Epstein-Barr virus (EBV) in tissues, because all infected cells express abundant EBER. Both the morphology and proportion of positive cells have to be assessed to determine the significance of the positive staining. Double staining (in situ hybridization for EBER combined with immunostaining for CD3 or CD79a) may be required to determine whether scattered EBER+ cells are of B or T lineage.
In situ hybridization for immunoglobulin light chain mRNA is an alternative to immunostaining for immunoglobulin light chain proteins. The advantages are that (1) the background is always very clean in contrast to the not uncommonly strong background seen in immunoglobulin immunostaining, because there is no circulating mRNA in the interstitium; and (2) only immunoglobulin-synthesizing cells are positive, and histiocytes and other cell types, which have absorbed immunoglobulin proteins into the cytoplasm, will not stain up (as with immunohistochemistry). The disadvantage is that the sensitivity is lower than immunohistochemistry; in general, this method is only suitable for detection of immunoglobulin light chain mRNA in plasma cells, as the amount of mRNA in lymphocytes is insufficient for detection.
With microarray technology, ordered arrays of oligonucleotides or other DNA sequences can be printed onto a solid support, permitting tens of thousands of genes to be analyzed simultaneously based on a small quantity of tissue. The main applications are identification of genetic alterations and assessment of gene expression profile. The technology has been successfully applied to the study of hematolymphoid neoplasms, such as use of gene expression profile to define or fine-tune tumor entities, identify normal counterparts, recognize clinically and biologically relevant subtypes within known tumor entities, and predict survival or response to treatment. The information derived from these studies has helped shape modern-day lymphoma classification. Gene expression profiling is currently not used on a routine basis in diagnostic work because of the complexities in technology, control standardization, result validation, and result interpretation. Nonetheless, the information gained from the many published studies can be utilized to select certain overexpressed or underexpressed targets of discriminatory value for analysis by reverse transcription PCR. Alternatively, a scaled-down version of gene expression profiling can be performed using the more robust NanoString platform.
High throughput nucleic acid sequencing technologies have allowed for the efficient survey of large numbers of genetic alterations in a single analysis. A rapidly evolving field, genomic analyses include targeted sequencing of hundreds of preselected genetic loci known to be mutated in tumors, complete sequencing of extracted mRNA (RNA sequencing), targeted sequencing of all known transcribed exons in the genome (whole exome sequencing), and complete sequencing of the tumor genome (whole genome sequencing). Each of these technologies has been applied to the analysis of lymphomas in the research setting, and targeted sequencing of preselected genetic loci has already been initiated for diagnostic use. Given its efficiency and dropping cost, high throughput sequencing may eventually replace select molecular and cytogenetic tests currently in use. The detection of genetic alterations in key signaling pathways may also guide targeted therapies.
Hodgkin lymphoma (HL) is characterized by the presence of Reed-Sternberg cells and their variants in an appropriate background of inflammatory cells. Both components of this definition must be satisfied for a diagnosis of HL to be made. Although HL was originally defined on morphologic criteria alone, immunohistochemical confirmation is advisable.
HL includes two biologically and clinically distinct entities: nodular lymphocyte predominant Hodgkin lymphoma (NLPHL) and classic Hodgkin lymphoma (CHL). NLPHL is a B-cell neoplasm, and CHL represents a neoplasm of “crippled” B cells.
HL accounts for 25% to 40% of all lymphomas in Caucasians, with one age peak in the second to third decades and another in the sixth decade. It is less common in Asians and in people of developing countries, where HL accounts for only 5% to 10% of all lymphomas. HL occurring in patients with acquired immunodeficiency syndrome (AIDS) exhibits a number of unusual features ( Box 21A.1 ).
Advanced stage at presentation (~90% stage III/IV)
Constitutional symptoms are common
Frequent involvement of extranodal and unusual sites. Bone marrow involvement is common (40%–50%)
Mediastinal involvement is less common
Noncontiguous pattern of spread
More likely to have aggressive subtypes of HL: mixed cellularity and lymphocyte depleted types more common (accounting for 66% of cases, compared with 29% in sporadic HL)
Background T cells mostly CD8+ cells instead of CD4+ cells
Aggressive clinical course and usually poor response to treatment. However, early stage disease treated aggressively is potentially curable
Much stronger association with EBV (80%–100%)
The World Health Organization (WHO) classification, which in turn is based on the prior Rye classification, has been widely used for many years and recognizes five categories of HL: NLPHL and four subtypes of CHL (nodular sclerosis, mixed cellularity, lymphocyte depleted, and lymphocyte rich) ( Table 21A.4 ). The commonest type of Hodgkin lymphoma is nodular sclerosis HL (NSHL), which accounts for more than 50% (up to 80%) of cases. Lymphocyte depleted HL (LDHL) is extremely rare, except in developing countries and immunocompromised hosts. Unclassifiable cases used to be placed in the mixed cellularity category, but the WHO classification recommends labeling such cases as HL, not classifiable . Most cases of anaplastic large cell lymphoma (ALCL) , Hodgkin–like have been shown to represent CHL, usually of the nodular sclerosis (NS) type.
| WHO | Rye | Lukes & Butler | |
|---|---|---|---|
| Nodular lymphocyte predominant | Lymphocyte predominance | Lymphocytic and histiocytic, nodular and diffuse | |
| Classic | Lymphocyte rich, classic | ||
| Mixed cellularity | Mixed cellularity | ||
| Mixed cellularity | |||
| Nodular sclerosis | Nodular sclerosis | Nodular sclerosis | |
| Lymphocyte depleted | Lymphocyte depletion | Reticular | |
| Diffuse fibrosis | |||
In the past, the lymphocyte predominant, nodular sclerosis, mixed cellularity (MC), and lymphocyte depletion (LD) subtypes have been shown to exhibit different clinical outcomes. These differences have been largely obliterated by modern therapy.
Although HL is regarded as a tumor distinct from NHL, there are overlaps and grey zone situations.
Almost any type of NHL (follicular lymphoma being the most common) can occur simultaneously with HL, in which case the term composite lymphoma may be used ; the two lymphoma components are often clonally related.
Patients with HL have an increased risk of developing NHL.
Some cases of chronic lymphocytic leukemia (CLL) have been observed to progress to HL. Patients with other lymphoma types, such as follicular lymphoma and mycosis fungoides, can also rarely develop HL subsequently.
There are cases in which a firm distinction between HL and NHL (particularly DLBCL) cannot be made despite exhaustive analysis by various techniques; these are termed, colloquially, grey zone lymphoma . To accommodate these difficult cases, the WHO classification has included a diagnostic category B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and CHL .
The Ann Arbor staging system has been widely used for staging of patients with HL and NHL ( Box 21A.2 ). The clinical stage is based on history, physical examination, radiologic studies, isotope scans, and laboratory tests and does not necessarily correlate with the pathologic stage. Histologic involvement of the spleen can occur in spleens weighing less than 200 g, while enlarged spleens are not always involved. Furthermore, subdiaphragmatic disease is found in one-third to one-fourth of patients with apparently clinical stage I/II disease.
| Stage I | Involvement of a single lymph node region (I) or a single extralymphatic a organ/site (IE) |
| Stage II | Involvement of two or more lymph node regions in the same side of the diaphragm (II), or |
| Localized involvement of an extralymphatic a organ and one or more lymph node regions on the same side of the diaphragm (IIE) | |
| Stage III | Involvement of lymph node regions on both sides of the diaphragm (III), which may also be accompanied by localized involvement of an extralymphatic a organ (IIIE) or involvement of the spleen (IIIS) or both (IIISE) |
| Stage IV | Diffuse or disseminated involvement of one or more extralymphatic a organs or tissues with or without associated lymph node enlargement |
a Extralymphatic organs are defined as those other than lymph node, spleen, thymus, Waldeyer ring, appendix, and Peyer patches.
Without symptoms listed below.
Systemic symptoms:
Unexplained fever 38°C
Unexplained weight loss 10% body weight in preceding 6 months
Night sweats
NLPHL, also known as nodular paragranuloma, accounts for only about 5% of all HLs. It most commonly occurs in children and young to middle-age adults ( Table 21A.5 ), with marked male predominance (M:F ratio 3 : 1). The patients usually present with a solitary enlarged peripheral lymph node, most commonly in the neck, groin, or axilla. Mediastinal lymph node involvement is very rare. Most patients have early stage disease (80% stage I or II), and systemic symptoms are uncommon. Bone marrow involvement is very rare and is associated with a poor prognosis.
| Nodular Lymphocyte Predominant HL | CLASSIC HL | ||||
|---|---|---|---|---|---|
| Lymphocyte-Rich Classic HL | Nodular Sclerosis HL | Mixed Cellularity HL | Lymphocyte Depleted HL | ||
| CLINICAL FEATURES | |||||
| Frequency | ~5% | ~5% | ~60% | ~25% | 1.5%–5% |
| Sex | M > F | M > F | F > M | M > F | M > F |
| Age | Usually young and middle-aged (median 35 years) | Slightly older (median age 43 years) | Usually early adulthood (median 28 years) | Any age (median 35 years) | Older (median 51 years) |
| Mediastinal involvement | Rare (7%) | Rare (15%) | Common (80%); contiguous pulmonary involvement is not uncommon | Sometimes (40%) | Rare |
| Sites of disease and clinical features | Lymphadenopathy usually of long duration, typically affecting single peripheral node. Clinically well. | Lymphadenopathy. | Mediastinal mass with supraclavicular node involvement is the commonest presentation. Often very bulky disease. Some (25%) cases show occult splenic disease. | Disease may involve many sites, but usually not particularly bulky. Spleen commonly affected. | Often widespread disease. Lymph nodes small or bulky. Fever, wasting, cytopenia, and hepatic dysfunction common. |
| B symptoms | Rare, ~10% | Rare ~10% | 42% | 35% | >50% |
| Stage I/II | 80% | 70-80% | 60% | 50% | Only 20% |
| PATHOLOGY | |||||
| Diagnostic RS cells | Rare or absent | Rare | Present | Usually easy to find | Usually easy to find |
| Major RS cell variants | L&H cells; rarely these cells can have large nucleoli resembling mononuclear or diagnostic RS cells | Mononuclear RS cells | Lacunar cells | Mononuclear RS cells | Mononuclear RS cells; pleomorphic RS cells |
| Appropriate inflammatory background | Lymphocytes and/or histiocytes only | Lymphocytes, plasma cells, histiocytes, eosinophils, and/or neutrophils | |||
| Other diagnostic features | Nodular pattern | More commonly nodular than diffuse | RS cells and variants tend to form aggregates, producing vague nodules. Dense fibrous bands traverse the lesion | RS cells and variants show a dispersed distribution, without nodular grouping | May show diffuse fibrosis |
| EBV association | <5% | ~40% | 10%–25% | ~75% | ~75% |
The natural history of NLPHL differs significantly from that of CHL. Relapse in the same or another nodal site is common (10%–32%), and may be delayed for many years (median time to relapse is ~4 years, which is longer than that of CHL) (see Table 21A.5 ) . Approximately 25% of the relapses are multiple. The histology in recurrent lesions remains remarkably constant, and relapses are often salvageable.
The prognosis of patients with stage I disease is excellent, with survival approaching the normal population. The 8-year HL-specific survival and failure-free survival rates for stage I and II are approximately 95% and 80%, respectively. On the other hand, the corresponding figures for stage IV are 41% and 24%, respectively. Patients with high stage disease who do not respond to therapy often die within 1 to 2 years.
When NLPHL is treated like CHL, the prognosis is excellent. However, mortalities from other causes, in particular second malignancy and cardiac disease, are more common than from the neoplasm. Furthermore, some patients treated with surgical excision alone have fared very well. Thus less intensive therapy, such as involved field radiation therapy, brief chemotherapy, rituximab, or a watch-and-wait strategy, is justified for early-stage disease.
The nodal architecture is usually obliterated, although a compressed rim of lymphoid tissue with reactive lymphoid follicles can be present. There are multiple, crowded, or well-separated large dark-staining nodules composed predominantly of small lymphocytes. When the nodules are back to back, they may show molding against one another ( Fig. 21A.7 ). The nodules are round, oval, or serpinginous. In 14% of cases, small germinal centers may be identified within some nodules. The nodules may appear mottled due to interspersed pink-staining histiocytes, while some nodules may be surrounded by wreaths of epithelioid histiocytes. Eosinophils and plasma cells are typically scanty or absent. The presence of sclerosis does not negate the diagnosis. A diffuse component can be present ( Fig. 21A.8 ).
 Typical example featuring multiple large dark-staining discrete nodules punctuated by pink foci occupied by histiocytes. The nodules are crowded and focally molded against one another. (B) Another example showing vague and focally coalescent nodules.")
 and histiocytes.")
The neoplastic cells used to be called lymphocytic and histiocytic (L&H) cells, but the WHO classification suggests renaming these cells lymphocyte predominance (LP) cells. They are mostly found within the nodules, but they can also be found between the nodules ( Fig. 21A.9 ). They characteristically possess lobulated nuclei with thin nuclear membrane, fine chromatin, and multiple small basophilic or eosinophilic nucleoli, earning them the nickname “popcorn cell.” Diagnostic Reed-Sternberg cells are not required for the diagnosis of NLPHL and, in fact, are often absent. When present, these cells exhibit the immunophenotype of L&H cells rather than diagnostic Reed-Sternberg cells ( Fig. 21A.10 ).
 resemble popcorn by virtue of the folded or lobated nuclear membranes. Nucleoli are typically not prominent. They are easiest to find within the nodules. Also note absence of plasma cells and eosinophils.")

Although the background lymphocytes are mostly bland-looking small lymphocytes, midsize lymphoid cells with more open chromatin and clear cytoplasm can form pale aggregates around the L&H cells in rare cases. Such cells are shown to be activated T lymphocytes ( Fig. 21A.11 ). In rare cases, there can be sheets or clusters of atypical T cells (midsize, often with irregular nuclei) between the nodules, mimicking peripheral T-cell lymphoma. These atypical T cells show no aberrant immunophenotype, and there is no clonal TR gene rearrangement.
 are scattered within these cellular aggregates.")
In extranodal sites, the pathologic and cytologic patterns are similar to those observed in lymph node. When the spleen is involved, there is often uniform involvement of the white pulp corpuscles (see Chapter 21B ).
Although NLPHL was previously thought to be accurately diagnosable by morphology alone, immunohistochemical studies are required to confirm the diagnosis. From the instructive study of von Wasielewski et al, only 76% of cases diagnosed on morphologic grounds as definite NLPHL by experts turn out to be bona fide NLPHL. Conversely, 12% of cases considered to be definite CHL turn out to represent NLPHL. Survival data show that immunohistochemical criteria are superior to morphologic assessment alone in the diagnosis of NLPHL, because cases categorized as NLPHL by immunohistochemistry have a significantly better survival than those not categorized as such (i.e. lymphocyte-rich CHL).
The most characteristic immunophenotypic feature of NLPHL is seen with the B–cell marker CD20: Positive large cells (L&H cells) occur within nodular aggregates of small B cells. The large cells stand out so clearly from the background small B lymphocytes because they are surrounded by a ring of nonstaining cells (T lymphocytes) ( Fig. 21A.12 ).
 The nodular pattern is accentuated because the nodules are rich in B lymphocytes. (Right) Higher magnification shows the diagnostic pattern of this neoplasm. Positively stained large cells occur within the nodules of small B cells, and they stand out strikingly because they are surrounded by rims of nonstaining T lymphocytes.")
L&H cells consistently display the full spectrum of B-lineage markers, such as CD20, CD79a, PAX5, PU.1, OCT2, and BOB.1 (see Fig. 21A.12 ). The immunohistochemical profile is contrasted with Reed-Sternberg cells of CHL in Table 21A.6 . CD30 positivity may be seen in up to 7% to 19% of cases, and CD15 is negative. EMA is positive in about half of the cases. Some studies have reported Ig light chain restriction in the L&H cells, with nearly all the cases expressing κ light chain.
| Nodular LPHL | Classic HL | |
|---|---|---|
| CD45RB | Positive, but often difficult to interpret because the L&H cells are tightly surrounded by small lymphocytes. | Reed-Sternberg cells and variants are negative, but interpretation is often difficult because these cells are submerged among lymphocytes. |
| CD20 | Positive. Many of the L&H (CD20+) cells are located within nodules of CD20+ small B cells. The positively stained L&H cells are typically surrounded by a ring of CD20- cells (T cells). | Negative or sometimes positive. If positive, usually in a heterogeneous pattern: some cells strongly positive, some weakly positive, some negative. |
| CD79a | Positive. | Negative or sometimes positive; the positivity rate is lower than for CD20. |
| CD30 | Usually negative (at most weak positive in some cells). | Positive. |
| CD15 | Negative. | Usually positive (~75%). |
| EMA | Usually positive. | Negative. |
| BCL6 | Positive (almost all neoplastic cells). | Usually negative. If positive, usually in <10% neoplastic cells. |
| EBV LMP1 | Negative. | Commonly positive (30%–60%). |
The L&H cells are positive for BCL6 and CD40, but not CD10 and fascin. Staining for MUM1 is either negative or weak.
In 27% of cases, the L&H cells express IgD. This subset of cases shows certain features distinct from the IgD–negative subset: younger median age (21 vs 44 years), striking male predominance (M:F ratio 23 : 1 vs 5 : 1), more frequent involvement of cervical nodes (56% vs 18%), and predominantly extrafollicular localization of the L&H cells.
The nodules are rich in polytypic small B lymphocytes with the phenotype of primary follicle or mantle zone B cells (IgM+, IgD+, CD10-, BCL6-). They are supported by CD21+/CD35+ FDC meshworks punctuated by multiple rarefied foci, which are inhabited by L&H cells and their rosetting T cells ( Fig. 21A.13A ). The rosetting CD3+ T lymphocytes usually express markers of follicular T-helper cells, namely PD1, CD57, BCL6, and CXCL13 (see Fig. 21A.13B and C ). In 27% of cases, the small lymphocytes within the nodules are mostly T cells rather than B cells ( Fig. 21A.14 ).
 CD21+ follicular dendritic cell meshworks can be demonstrated in the nodules. Typically the meshworks are punctuated by multiple holes. (B) Staining for CD3 demonstrates rosettes of T cells around the L&H cells. (C) Only some but not all of the rosetting T cells are CD57 positive.")

The small lymphocytes between the nodules and in the diffuse areas are predominantly T cells (CD3+), with very few small B cells. The number of CD57+ small lymphocytes is usually sparse in the diffuse areas.
Clonal IG gene rearrangements are usually not detected in NLPHL tissue samples either by Southern blot or standard PCR technique. However, using PCR to analyze microdissected L&H cells, clonal rearrangements of IG genes can be demonstrated, confirming the B-cell nature of the neoplastic cells. IG mRNA transcripts can be detected in the L&H cells. The presence of somatic hypermutations in the IG gene and intraclonal diversity, aberrant somatic hypermutations (targeting PIM1, PAX5, RhoH/TTF, MYC genes), presence of BCL 6 gene rearrangement in 48% of cases, and the histologic and immunohistochemical findings (presence of nodules with features of altered follicles, presence of discrete FDC meshworks, expression of B–lineage markers and BCL6) strongly suggest a relationship of NLPHL with follicular center B cells. However, it is not a variant of follicular lymphoma, because BCL2 gene rearrangement is lacking.
Specific cytogenetic abnormalities have not been found. Comparative genomic hybridization reveals a high number of genomic imbalances (mean 10.8 per case, including gains and losses of multiple chromosomes or chromosome segments). The L&H cells generally do not harbor EBV ; the rare EBV-positive cases appear to have a more aggressive behavior.
Approximately 3% to 17% of cases of NLPHL are complicated by a DLBCL, which is discovered either at initial presentation or subsequently after an interval of 0.5 to 24 years (median 1 year). The DLBCL is composed of confluent sheets of large cells resembling L&H cells, centroblasts, or immunoblasts ( Fig. 21A.15 ), which express B-lineage markers and often monotypic Ig. It is unclear whether the large cell lymphoma represents genuine NHL or merely tumorous overgrowth of L&H cells. A clonal relationship between the DLBCL and NLPHL is demonstrated in some but not all cases. EBV does not play a role in the transformation. While earlier studies suggested that DLBCL arising in NLPHL is associated with a favorable prognosis, more recent studies have shown a prognosis comparable to de novo DLBCL.

Rare cases of peripheral T-cell lymphoma found concurrently with or subsequently to a diagnosis of NLPHL have been reported. A few cases of NLPHL associated with CHL have also been described.
Progressive transformation of germinal centers (PTGC). PTGC is a reactive lesion characterized by large dark-staining follicles scattered in a background of usual reactive follicles ( Fig. 21A.16 ). The large follicles are several times the size of the background follicles. Most have thick mantles (IgD+), which surround one or several irregular-shaped germinal centers with blurred outlines. Some are composed mostly of small lymphocytes, sprinkled with germinal center cells. A pathogenetic relationship between PTGC and NLPHL has been proposed. They may coexist in the same lymph node; some patients with NLPHL on follow-up develop PTGC, and a small percentage of patients with PTGC subsequently develop NLPHL. Thus when recurrent lymphadenopathy occurs in a patient with NLPHL, the node must be biopsied to determine whether the patient has tumor recurrence, PTGC, or another unrelated pathologic process. In all cases of florid PTGC, early NLPHL should be excluded by searching for discrete expansile coalescent follicles and L&H cells. T-cell rosettes around large CD20+ cells are a constant feature of NLPHL but are rare in PTGC. Residual germinal centers are common in the large nodules of PTGC but very rare in those of NLPHL. In difficult cases, immunostaining for PAX5, BCL6, or OCT2 is particularly helpful for highlighting L&H cells (large cells with irregular nuclear contours). See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.

Follicular lymphoma. Rare cases of follicular lymphoma may show architectural features mimicking NLPHL. In contrast to the latter, the lymphoid cells that comprise the nodules are follicular center cells (often rich in centrocytes with triangular or elongated nuclei, and positive for CD10 and BCL6) rather than small lymphocytes and L&H cells. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLLSLL). The predominance of small lymphocytes in NLPHL may lead to a mistaken diagnosis of CLL/SLL. However, CLL/SLL is very rare below the age of 40 years, and the small B lymphocytes coexpress CD5 and show light chain restriction.
CHL, particularly nodular lymphocyte-rich CHL. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Peripheral T-cell lymphoma, follicular variant.
Nodular B-cell–rich FDC sarcoma.
Currently it is not possible to recognize a pure diffuse LPHL, due to the marked overlap in clinicopathologic features between NLPHL and T-cell/histiocyte-rich large B-cell lymphoma ( Fig. 21A.17 ). When a neoplasm considered to be a likely candidate for diffuse LPHL is found to have a minor nodular component on reticulin stain or immunostaining for B-cell markers, it should be diagnosed as NLPHL. If no nodular component is identified upon thorough sampling, the case should be given the diagnostic label T-cell/histiocyte-rich large B-cell lymphoma instead. This also means that a definitive diagnosis is practically impossible in small biopsies .

The presence of a significant diffuse component in NLPHL is associated with high stage disease (stage III/IV) and a higher relapse rate.
Most patients with CHL present with lymphadenopathy. The enlarged nodes are often multiple, rubbery to firm, nontender, and matted. Extranodal disease in the absence of lymph node involvement is uncommon. NSHL differs clinically from other types of CHL in that there is a female predominance and frequent involvement of the mediastinum. The major clinical features of the various subtypes of HL are listed in Table 21A.5 .
CHL spreads predominantly in a contiguous pattern, as evidenced from the findings in staging laparotomy and the success of extended field radiotherapy in patients with early stage HL. HL begins as a unifocal disease and spreads to the next group of lymph nodes in a manner similar to carcinomas via lymphatics, including retrograde flow. Spread to the spleen is thought to occur following paraaortic and splenic hilar lymph node involvement. Once the spleen is involved, bloodborne spread can take place, such as to the liver and bone marrow. This theory, however, fails to explain some cases of skip involvement, bilateral cervical/axillary involvement with no intervening nodes, and splenic involvement in the absence of afferent lymphatics in the spleen. This theory also does not hold for HL occurring in patients with AIDS.
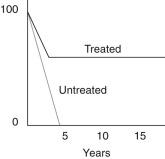
The historic 5-year survival rate of untreated patients with CHL was less than 5% ; however, with currently available therapy, a high proportion of patients can be cured. Between 1960 and 1992, the 5–year survival of patients with HL in the United States rose from 40% to 82%, representing one of the greatest triumphs of modern cancer therapy. The treatment modality has to be individualized based on the clinical stage and various prognostic factors, to minimize treatment toxicity while maintaining a high cure rate. Treatment usually includes chemotherapy, involved field radiation, or a combination. The standard chemotherapy regime is ABVD (adriamycin, bleomycin, vinblastine, and dacarbazine), and risk-adapted approaches have helped deescalate therapy in low-risk patients and intensify treatment for high-risk patients.
The great majority of patients can be brought into remission. Approximately 30% of these responders, especially those with more advanced disease, will suffer a relapse, usually within 2 to 3 years. Salvage therapy can often successfully reinduce remissions. Novel targeted therapies, such as CD30-targeting antibody-drug conjugates (e.g., brentuximab) and blockade of the PD1/PDL1 pathway (nivolumab and pembrolizumab), hold promise in inducing remissions for patients with relapsed disease.
In long-term survivors of HL, second malignancies (such as acute leukemia and solid cancers) related to treatment are a significant concern.
The neoplastic cells in HL are Reed-Sternberg cells and their variants. Diagnostic Reed-Sternberg cells are necessary but not sufficient for the diagnosis of CHL, because cells resembling Reed-Sternberg cells can be found in NHLs, reactive lymphadenopathies (including infectious mononucleosis), and other malignant tumors. To render a diagnosis of CHL, both of the following features must be satisfied: (1) presence of Reed-Sternberg cells or variants and (2) presence of an inflammatory cellular background appropriate for the specific subtypes of HL (see Table 21A.5 ).
Diagnostic Reed-Sternberg cells are large cells (20–30 µm) with morphologic hallmarks of polyploidy (double, multiple, or multilobed nuclei); they do not always show the mirror-image double nuclei often portrayed in textbooks ( Figs. 21A.18 and 21A.19 ). The nuclear membrane is thick, the chromatin is vesicular, and the nucleolus is large, eosinophilic, and inclusion-like (larger than an erythrocyte), often surrounded by a halo. The cytoplasm is lightly eosinophilic to amphophilic. Diagnostic Reed-Sternberg cells occur in all subtypes of CHL.
 in classic Hodgkin lymphoma. Note the huge eosinophilic inclusion-like nucleoli. The background is composed of nonactivated small lymphocytes mixed with some plasma cells and eosinophils.")

Mononuclear Reed-Sternberg cells are identical to diagnostic Reed-Sternberg cells except that they possess a single round or oval nucleus ( Fig. 21A.20 ). They may be found in the various subtypes of CHL.
 . The large cells have central large nucleolus. A diagnostic Reed-Sternberg cells is seen in the left field.")
Lacunar cells are characteristic of NSHL. They possess round or lobated nuclei, vesicular chromatin, and multiple distinct small to midsize nucleoli. The voluminous cytoplasm is pale and retracted, and thus the tumor cells appear to lie within lacunae ( Fig. 21A.21 ). This diagnostic feature is, however, an artifact of formalin fixation.

Pleomorphic Reed-Sternberg cells have large bizarre polyploid nuclei. Although typical of LDHL, they can also occur in NSHL and MCHL.
Mummified cells are large necrobiotic cells in which the nuclear details are no longer discernible and the cytoplasm is deeply eosinophilic ( Fig. 21A.22 ). They are a common finding in CHL but are not diagnostic by themselves.

Although the requirement to identify diagnostic Reed-Sternberg cells for the diagnosis of HL is emphasized in the literature and textbooks, there is no need to spend undue effort in looking for them. In some cases of CHL, diagnostic Reed-Sternberg cells are sparse, or most of the neoplastic cells more closely resemble those of conventional large cell lymphoma. In practice, in the presence of an appropriate inflammatory background, as long as a typical immunophenotype can be demonstrated and at least some large cells (mononuclear, bilobed, or multilobed) exhibit very large nucleoli, a diagnosis of CHL can be made ( Fig. 21A.23 ).

Historically, the histogenesis of Reed-Sternberg cells was controversial, with the histiocyte, interdigitating dendritic cell, FDC, myeloid cell, and lymphocyte all implicated as the probable cell of origin. However, it is now widely accepted that Reed-Sternberg cells in CHL are of B lineage in nearly all cases, as evidenced by expression of B–cell–specific activator protein PAX5 and sometimes CD20, presence of IG gene rearrangement, and a pattern of genome expression compatible with B cells. Reed-Sternberg cells show a global downregulation of the B-cell gene expression program, which is unique among B-cell neoplasms. They show features of germinal center or postgerminal center B lymphocytes, but are crippled—that is, despite presence of IG gene rearrangement they fail to produce functional IG mRNA transcripts and Ig proteins, and furthermore often lack expression of B-cell–specific antigens. The discrepancy may be caused by crippling mutations of the IG genes (~25% of cases) or downregulation and disruption of the B-cell transcription factors, such as OCT2, BOB.1, and PU.1. Current understanding holds that these cells are rescued from apoptosis through activation of the NFkB and/or JAK/STAT signaling pathway(s), although the precise transforming events remain under investigation and likely have not been fully catalogued.
The rare existence of a T-cell type of CHL, whereby the Reed-Sternberg cells show immunohistochemical and molecular evidence of T lineage differentiation, remains an unsettled issue.
In CHL, the Reed-Sternberg cells and variants are often CD45RB-, CD30+, CD15+, CD20-, PAX5+ (see Table 21A.6 ). CD30 is positive in nearly 100% of cases and often shows perinuclear accentuation, representing staining of the Golgi complex. CD15 staining, demonstrable in nearly 75% of cases, should take the form of a paranuclear globule with or without cell membrane staining ( Fig. 21A.24 ). Lack of CD15 expression is associated with an older age group, male predominance, higher stage disease, and more commonly MC than NS histology. The intensity of staining for PAX5 is typically weak to moderate compared with the surrounding B cells. Although CD20 is usually negative, about 20% of cases show CD20 immunoreactivity in Reed-Sternberg cells, typically with a heterogeneous staining pattern (some cells strongly positive, some weakly positive, and some negative) ( Fig. 21A.25 ).
 The Reed-Sternberg cells show cell membrane and Golgi staining for CD30. (Right upper) CD15 staining highlights the nodular grouping of neoplastic cells in this example of nodular sclerosis Hodgkin lymphoma. (Right lower) CD15 staining in Reed-Sternberg cells should be in the cell membrane or Golgi zone.")
 In some cases, some large neoplastic cells show weak or strong staining for CD20, while some are negative. (Right) The background is rich in CD3+ T cells, while the neoplastic cells are negative.")
The Reed-Sternberg cells are often positive for HLA–DR, CD25, CD40, MUM1, fascin, and TRAF1. BCL6 is expressed in a small percentage of tumor cells in 30% of cases.
Reed-Sternberg cells commonly exhibit staining for Ig, but both κ and λ light chains are demonstrable in the same cell, indicating absorption of Ig from the surrounding rather than cellular synthesis. The B–cell–associated transcription factors PU.1, OCT2, and BOB.1 are usually all negative, or only one or two of them are expressed. Staining for T-lineage markers, such as CD2, CD3, CD4, CD5, and CD8, has been reported in up to 5% of cases, usually only a single marker in a proportion of tumor cells.
The background small lymphocytes are mostly T cells, with CD4+ cells greatly outnumbering CD8+ cells (see Fig. 21A.25 ). The T cells commonly express cytotoxic molecules, but do not express CD57. Networks of FDCs enmeshing the neoplastic cells can be found in some cases.
CHL usually lacks rearrangements of the IG genes on Southern blot analysis, except in cases with a high content of Reed-Sternberg cells or fractions enriched with Reed-Sternberg cells. When PCR is used to detect clonal IG gene rearrangement in whole tissue, positive results are obtained in 23% to 50% of cases, and the rate is higher for cases showing CD20 immunoreactivity. TR gene rearrangements are usually not detected.
PCR analysis on single neoplastic cells microdissected from the tissues shows that IG gene is rearranged and exhibits somatic hypermutations. Comparison of the DNA sequences of the rearranged IG gene of the different cells obtained from the same case confirms the clonal nature of the neoplastic cells. There are commonly also aberrant somatic hypermutations targeting genes expressed in germinal center cells ( PIM1, PAX5, RhoH/TTF, MYC ).
Numerical chromosomal abnormalities are common in CHL, but there are no specific nonrandom chromosomal abnormalities. However, rearrangement of the locus encoding the MHC class II transcriptional activator CIITA occurs in up to 15% of cases; the functional consequence is downregulation of HLA class II expression and upregulation of molecules that suppress an antitumor immune response, such as PD-L1 and PD-L2. Amplification of the genetic locus at 9p24.1, the region encoding PD-L1 and PD-L2 , is also observed in a subset of CHL ; these cases are more likely to have advanced stage disease and a worse prognosis.
Gene expression profiling studies show that Reed-Sternberg cells exhibit decreased mRNA levels for nearly all established B-lineage–specific genes, affecting multiple components of signaling pathways active in B cells, including B-cell receptor signaling. Molecular groups with different clinical outcome can be identified: the good prognosis group overexpresses genes involved in apoptotic induction and cell signaling, while the poor prognosis group is characterized by upregulation of genes involved in fibroblast activation, angiogenesis, extracellular matrix remodeling, cell proliferation, and downregulation of tumor suppressor genes.
In Western countries, 40% to 50% of CHL are associated with EBV, suggesting a pathogenetic role of the virus. The association is strongest for MC type (>60%) and LD type, and is weaker for NS type (<30%). There is a stronger association with EBV for CHL occurring in the head and neck region, and CHL occurring at the extremes of life. In Asian countries, the association rate is approximately 60%. In patients from developing countries and patients with AIDS, there is a near 100% association with EBV, suggesting that EBV plays an even more important role in the genesis of CHL arising in an immunocompromised host.
EBV is present in a clonal episomal form, indicating that EBV infection preceded expansion of the neoplastic clone. Among the three main patterns of EBV gene expression (latency) in EBV–associated tumors ( Table 21A.7 ), CHL characteristically exhibits type II latency, with EBV latent membrane protein-1 (LMP1) expression.
| EBV LATENCY | |||
|---|---|---|---|
| Type I | Type II | Type III | |
| EBNA1 | + | + | + |
| EBNA2-6 | − | − | + |
| LMP1 | − | + | + |
| Lymphoma types exhibiting the EBV latency | Burkitt lymphoma |
|
|
Many different techniques can be used to demonstrate the presence of EBV. PCR has the drawback that a positive result may be obtained even if EBV is merely present in bystander lymphocytes. In situ hybridization for EBV encoded early RNA (i.e., EBER) is a highly sensitive and relatively simple method for demonstration of EBV at the cellular level: Reed-Sternberg cells and occasional small lymphocytes show positive nuclear labeling. Immunostaining for EBV LMP1 is a simple alternative for demonstration of EBV in CHL, because this protein is consistently and strongly expressed by Reed-Sternberg cells in EBV+ cases. The staining should be in the cell membrane and Golgi zone.
Although the prognostic implication of EBV positivity in CHL has been controversial, many studies have shown that EBV-positive CHLs occurring in patients over the age of 50 years are associated with a worse prognosis.
The prototypic patient is a young adult (with female predilection) presenting with bulky mediastinal tumor mass, sometimes accompanied by supraclavicular lymphadenopathy (see Table 21A.5 ). Local or distant relapses can occur, but the disease runs true in that the HL always remains as NS type in the relapses, even though the proportion of neoplastic cells can increase. With modern therapy, 5-year progression-free survival is over 80%.
A diagnosis of NSHL takes precedence over other histologic types of HL whenever it is present, even focally. The lymph node shows focal, partial, or total involvement. Typically the capsule is thickened, with multiple broad birefringent and vascularized collagenous bands extending into the parenchyma, resulting in the formation of multiple nodules ( Fig. 21A.26 ). However, sclerosis can be minimal in some cases; it has been suggested that a minimum of a single polarizable fibrous band is required for the diagnosis. There is a tendency for the neoplastic cells to form loose aggregates, resulting in a vaguely nodular pattern (see Fig. 21A.26B ). The centers of the nodules may show necrosis and/or suppuration (see Fig. 21A.26 ). With time, the nodules may undergo obliterative sclerosis.
 Typical example showing extensive sclerosis and nodule formation. Note the marked fibrous thickening of the capsule. (B) A broad fibrous band extends from the thickened capsule into the parenchyma. Central necrosis is evident within the nodule of lacunar cells; this is a common finding.")
Within the nodules, lacunar cells are admixed with variable numbers of small lymphocytes, plasma cells, eosinophils, neutrophils, and histiocytes. In some cases, the background cells can be comprised almost exclusively of small lymphocytes (see Figs. 21A.21 and 21A.23 ). The lacunar cells occur singly, in aggregates or in sheets. They can appear monomorphous or can show considerable pleomorphism, and their nucleoli can be small or prominent ( Fig. 21A.27 ; also see Figs. 21A.21 and 21A.23 ). Diagnostic Reed-Sternberg cells are infrequent.
 Lacunar cells occur in a background rich in small lymphocytes, consistent with BNLI grade 1. (B) In this example, bizarre lacunar cells are found in most nodules in the absence of lymphocyte depletion, consistent with BNLI grade 2.")
The syncytial variant of NSHL is characterized by neoplastic cells forming cohesive cellular aggregates and sheets ( Fig. 21A.28 ). The neoplastic cells sometimes do not show the typical cytologic features of lacunar cells, but more resemble those of large B-cell lymphoma. Necrosis, sometimes geographic in shape, is common. This variant may be mistaken for metastatic carcinoma, metastatic melanoma, or large cell lymphoma. It is associated with advanced stage disease at presentation.
 The neoplastic cells occur in sheets, with few or no lymphocytes in between, consistent with BNLI grade 2. It is difficult to distinguish this lesion from a large cell lymphoma. (Right) Immunostaining for CD30 highlights the sheets of tumor cells.")
The cellular phase of NSHL is a more controversial entity. It refers to cases showing a nodular pattern and conspicuous lacunar cells, but lacking fibrous bands. It is appropriate to include this entity under NSHL rather than MCHL, because further biopsies often reveal features of typical NSHL.
The British National Lymphoma Investigation (BNLI) has found grading of NSHL to be of prognostic importance ( Table 21A.8 ). NSHL is categorized as either grade 1 (NS1) or grade 2 (NS2). For NS2, apparently effective irradiation of local disease may be followed by local recurrence, and both this and late abdominal relapse more often prove refractory to salvage treatment. The outcome of NS2 can be improved with more intensive treatment. The usefulness of this grading system has been confirmed by some studies, but not others. One possible explanation for these conflicting results is that aggressive therapy may obliterate differences in outcome. The other explanation is that, since the prognostic difference between the grades is observed only in advanced stage disease but not early and intermediate stage, studies including only or predominantly patients with early stage disease may not demonstrate a difference.
| NSHD Grade 1 (NS1) | NSHD Grade 2 (NS2) | |
|---|---|---|
| Presence of B symptoms | 29% | 51% |
| 5-year survival: | ||
| Stage I/II | 92% | 74% |
| Stage III/IV | 77% | 55% |
| Overall | 83% | 66% |
| Complete response rate | 84% | 66% |
| Disease-free survival at 5 years | 54% | 37% |
A lesion is assigned to the grade 2 category when any of the following features is present ( Fig. 21A.29 ; see also Figs. 21A.27B and 21A.28 ):
25% of cellular nodules contain numerous bizarre and highly anaplastic Hodgkin cells, without depletion of lymphocytes.
25% of cellular nodules show lymphocyte depletion, irrespective of whether the neoplastic cells appear uniform or anaplastic.
80% of cellular nodules show a fibrohistiocytic pattern (many histiocytes and fibroblasts, with relatively few lacunar or Reed-Sternberg cells) (see Fig. 21A.29 ).

Overall, about 80% of all cases fall into the grade 1 category. Doubtful or borderline cases are also classified as grade 1. The syncytial variant corresponds to grade 2 . The fibroblastic variant, which had been associated with a shorter relapse-free survival in a previous study, corresponds to the fibrohistiocytic type of NS2.
Necrotizing lymphadenitis and cat-scratch disease. Some cases of NSHL may exhibit such extensive coagulative or suppurative necrosis that their neoplastic nature may be obscured, leading to an erroneous diagnosis of necrotizing lymphadenitis or cat-scratch disease. One should always be on the alert for NSHL in lymph nodes obtained from the mediastinum or lower neck. The best place to search for the neoplastic cells is around the necrotic foci, although distinction from histiocytes can be difficult because the nucleoli of the lacunar cells can be small. Immunostaining for CD30 is of great help in highlighting this population.
Sclerosing inflammation or sclerosing mediastinitis. Small biopsies of NSHL showing significant obliterative sclerosis are likely to be mistaken for sclerosing inflammation. Serial sections should be examined for lacunar cells and Reed-Sternberg cells, and repeat biopsy should be performed if HL is suspected clinically and yet the histologic findings are nondiagnostic.
Undifferentiated pleomorphic sarcoma. The fibrohistiocytic-fibroblastic variant of NSHL may be mistaken for a soft tissue neoplasm. However, the spindle cells lack definite atypia. More careful search will reveal occasional lacunar cells and Reed-Sternberg cells.
Metastatic cancer. The syncytial variant of NSHL can be difficult to distinguish from metastatic carcinoma or melanoma. The presence of spindle-shaped tumor cells, prominent sinusoidal pattern, significant nuclear pleomorphism, and emperipolesis of neutrophils in tumor cells should raise a strong concern for metastatic carcinoma. The diagnostic problem can easily be settled by immunohistochemistry.
Large cell lymphoma. Distinction of the syncytial variant of NSHL from large B-cell lymphoma can be very difficult. The presence of foci of typical NSHL favors the former diagnosis. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
There are no distinctive clinical features (see Table 21A.5 ). Besides involvement of cervical and supraclavicular lymph nodes, there is a tendency to involve intraabdominal nodes. With modern therapy, 5-year progression-free survival is over 80%. This HL type can progress with time to LDHL.
The lymph node is partially or diffusely involved. In a background of small lymphocytes, plasma cells, eosinophils, neutrophils, and histiocytes, mononuclear and diagnostic Reed-Sternberg cells are readily found ( Fig. 21A.30 ; see also Figs. 21A.18 and 21A.20 ). The neoplastic cells are evenly dispersed and do not exhibit nodular grouping as in NSHL. Not all of them show the typical features of Reed-Sternberg cells and variants; some may simulate immunoblasts or show features intermediate between immunoblasts and mononuclear Reed-Sternberg cells. Some cases may be rich in epithelioid histiocytes, mimicking the lymphoepithelioid variant of peripheral T-cell lymphoma (so-called Lennert lymphoma). Focal necrosis with secondary fibrosis may be found. The latter lacks the refractile, banded quality of the fibrous tissue seen in NSHL.

Although the lymphocytes in the background of HL are classically described as small round lymphocytes, nonactivated, and lacking blasts, they can in fact have slightly larger and irregular nuclei in some cases (see Fig. 21A.20 ). Small numbers of activated large blastic cells are also acceptable.
Peripheral T-cell lymphoma. In peripheral T-cell lymphoma, the smaller lymphoid cells usually show atypia and irregular nuclear foldings, and there is often a continuous spectrum of atypical small, midsize, and large cells. The presence of clear cells also favors a diagnosis of T-cell lymphoma. Immunohistochemical studies are required to confirm the diagnosis.
T-cell/histiocyte-rich large B-cell lymphoma. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” later in the chapter.
Reactive immunoblastic proliferations, such as infectious mononucleosis. Cells indistinguishable from Reed-Sternberg cells can occur in reactive immunoblastic proliferations. However, the background is rich in activated lymphoid cells and immunoblasts, which never occur in abundance in HL.
Interfollicular Hodgkinoid lymphadenitis. A reactive condition featuring interfollicular proliferation of lymphocytes, immunoblasts, plasma cells, eosinophils, and epithelioid histiocytes may mimic HL. The immunoblasts are, however, not as large as mononuclear Reed-Sternberg cells, their nucleoli tend to be basophilic rather than eosinophilic, and their cytoplasm is basophilic/amphophilic rather than eosinophilic. They react with B–cell or T–cell antibodies but not CD15.
Metastatic carcinoma or melanoma.
Castleman disease, plasma cell type. Rare cases of MCHL may present with a histologic picture indistinguishable from plasma cell–type Castleman disease. The component of HL is often subtle and discovered only on active scrutiny.
Lymphocyte-rich classic HL (LRCHL) is rare, accounting for only 4% of all HLs. Like NLPHL and in contrast to NSHL or MCHL, patients with LRCHL often have early stage disease, infrequent B symptoms, and uncommon occurrence of mediastinal involvement. However, the median age is higher than that of NLPHL (see Table 21A.5 ). The survival rate of LRCHL is slightly better than that of other subtypes of CHL, but is similar to that of NLPHL (8-year HL-specific survival 87% vs 95%; 8-year failure-free survival 75% vs 74%). Nonetheless, compared with NLPHL, relapses are less frequently multiple (5% vs 27%), and survival after relapse is worse.
LRCHL is characterized by Reed-Sternberg cells and variants singly scattered in a background rich in small lymphocytes and with sparse or absent granulocytes ( Fig. 21A.31 ). The growth pattern can be nodular (much more common) or diffuse.

In the nodular variant (N-LRCHL), large crowded nodules are evident at low magnification, architecturally very similar to NLPHL. The nodules are composed predominantly of small lymphocytes, but some can have small germinal centers. Reed-Sternberg cells and variants are scattered within, and sometimes between, the lymphoid nodules ( Fig. 21A.32 ). In some cases, within the lymphoid nodules, the Reed-Sternberg cells and variants are surrounded by aggregates of pale-staining midsize cells (T cells). There are few admixed plasma cells and neutrophils.
 Closely packed, dark-staining, large lymphoid nodules characterize the low magnification appearance of this form of Hodgkin lymphoma. (B) Some nodules can be formed by follicles with broad mantles, in which are scattered Reed-Sternberg cells.")
On immunohistochemical evaluation, the large cells show the typical immunophenotype of Reed-Sternberg cells. The small lymphocytes constituting the nodules are mostly CD20+ B cells with IgD expression. The neoplastic large cells are often rosetted by CD3+ T cells, which exhibit a T follicular helper cell phenotype (PD1+, CD57+/-) in about half of the cases. The CD21+ FDC meshworks typically show multiple small cavities populated by the large neoplastic cells and their surrounding T cells. The interfollicular zone is rich in T cells. Molecular analysis shows mutated rearranged IG genes without significant intraclonal diversity, similar to the pattern seen in other types of CHL.
LDHL is extremely rare in Western populations, and it occurs predominantly in developing countries or in the setting of immunosuppression (such as AIDS). There is marked male predominance. Patients often have high stage disease and B symptoms.
There are two recognized morphologic types :
Diffuse fibrosis type. In a background of disorderly distributed reticular fibrous (nonbirefringent) and amorphous proteinaceous material, there is striking depletion of all cellular elements. Diagnostic Reed-Sternberg cells are rare ( Fig. 21A.33 ). Necrosis may be prominent.

Reticular type. The lesion is hypercellular, with abundance of diagnostic and pleomorphic Reed-Sternberg cells ( Fig. 21A.34 ). Mature lymphocytes are sparse, as are plasma cells, eosinophils, histiocytes, and neutrophils. There may be disorderly, nonbirefringent fibrous tissue. Necrosis is common.

With better delineation of the various types of NHL, a diagnosis of LDHL is rarely made nowadays. Reassessment of cases so diagnosed in the past has shown that most cases are reclassifiable as (1) NSHL with lymphocyte depletion, and (2) large cell NHL, usually of anaplastic large cell, peripheral T-cell, or T-cell/histiocyte-rich large B-cell types. Very few cases represent bona fide examples of LDHL. Regardless of the number of Reed-Sternberg cells and variants, the case should be classified as NSHL if lacunar cells predominate and there is associated sclerosis.
The prognosis of patients with LDHL used to be very poor, but 5-year progression-free survival rate of 71% is obtainable in recent years with appropriate chemotherapy.
HL of various subtypes can exhibit predominantly interfollicular growth. The accompanying reactive follicular hyperplasia may be so prominent as to distract the pathologist from the subtle component of HL.
An abundance of foamy histiocytes in some cases of HL may lead to confusion with xanthogranulomatous inflammation or lipid-storage disease.
Some cases of HL may be associated with coalescing small clusters of epithelioid histiocytes, mimicking the pattern of Lennert lymphoma. Most belong to the MC category.
Very rarely, HL is localized entirely or predominantly within clusters of reactive monocytoid B cells, and thus the diagnosis can be missed ( Fig. 21A.35 ).
, there are scattered large Reed-Sternberg cells.")
Recurrent HL after therapy may show very atypical histologic features. The cellular proliferation may appear rather monomorphous, rendering distinction from NHL difficult. Mass lesions that sometimes persist after treatment of HL may be composed only of hyalinized tissue with no evidence of residual disease.
Although once popular, staging laparotomy is rarely performed nowadays. If a histologic diagnosis of CHL has already been firmly established from lymph node biopsy, less stringent criteria can be used for the diagnosis of involvement in other tissues. As long as atypical large cells with the typical immunophenotype are found in an appropriate background, involvement by HL can be diagnosed even in the absence of diagnostic Reed-Sternberg cells.
In the spleen, the number of abnormal nodules should be counted, because the presence of five or more nodules is associated with a worse prognosis. HL first affects the white pulp in a periarteriolar location or in the marginal zone; the tumor gradually expands to encroach upon the lymphoid follicles and red pulp, forming coalescent nodules (see Chapter 21B ).
In the liver, the portal tracts are involved, with gradual encroachment on the parenchyma. In the marrow, focal or diffuse fibrosis with a nonspecific infiltrate of lymphocytes, histiocytes, eosinophils, and plasma cells is also suggestive of involvement even in the absence of atypical cells. Marrow eosinophilia is a form of reaction sometimes seen in patients with CHL; this alone should not be considered marrow involvement by CHL.
Clinical factors. High stage disease is an unfavorable prognostic factor. The male sex, advanced age, B symptoms, large tumor load, and failure to achieve complete remission are associated with a worse prognosis.
Pathologic features. The prognostic significance of histologic typing has practically disappeared with the availability of modern therapy. Vascular invasion is associated with a worse prognosis. Histologic grading of NSHL is of prognostic significance according to some but not all studies (see “Nodular Sclerosis Hodgkin Lymphoma” earlier in the chapter).
Immunophenotype. Lack of expression of CD15 or expression of CD20 has been correlated with a less favorable outcome. Presence of FDC meshworks among the neoplastic cells is associated with a more favorable prognosis.
Microenvironment. By immunohistochemistry or gene expression profiling, high numbers of tumor-associated macrophages are associated with poor overall survival. This may be due in part to a paracrine survival mechanism between the Reed-Sternberg cells and local macrophages. Similarly, the immunoregulatory glycan-binding protein, galectin-1, produced by Reed-Sternberg cells, suppresses antitumor directed T-cell immunity, and its expression predicts poor overall and event-free survival. However, standardized scoring systems for tumor-associated macrophages and expression of immunoregulatory molecules in CHL have yet to be implemented in standard practice .
For a long time, there was no unified classification of NHLs, creating confusion in the literature. The situation was brought to order with the introduction of the Revised European-American Lymphoma (REAL) Classification by the International Lymphoma Study Group in 1994. The classification emphasized biologic entities, departing from a pure morphologic approach in classification. It has gained worldwide acceptance, and many studies have demonstrated its clinical relevance ( Table 21A.9 ). The classification has since been incorporated in the 2001 WHO classification (3rd edition), which was updated in 2008 (4th edition) and further updated in 2017 (revised 4th edition) ( Boxes 21A.3 and 21A.4 ). There is great emphasis on the site of disease and specific genetic alterations in the definition of the disease entities, with several new categories in the most recent revision based on molecular findings.
| Histologic Type of NHL | Frequency | Median Age (yr) | M:F Ratio | STAGE DISTRIBUTION | Bulky Disease ≥10 cm | Marrow Involvement | % With Poor IPI Score | 5-Year Overall Survival a | 5-Year Failure-Free Survival a | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | |||||||||
| Lymphoblastic lymphoma (T or B lineage) | 2% | 28 | 1.8 : 1 | 0% | 11% | 14% | 75% | 32% | 50% | 26% | 26% | 24% |
| CLL/SLL | 6% | 65 | 1.1 : 1 | 4% | 5% | 8% | 83% | 13% | 72% | 13% | 51% | 25% |
| Lymphoplasmacytic lymphoma | 1% | 63 | 1.1 : 1 | 7% | 13% | 7% | 73% | 25% | 73% | 15% | 59% | 25% |
| Mantle cell lymphoma | 6% | 63 | 2.8 : 1 | 13% | 7% | 9% | 71% | 25% | 64% | 23% | 27% | 11% |
| Follicular lymphoma | 22% | 59 | 0.7 : 1 | 18% | 15% | 16% | 51% | 28% | 42% | 7% | 72% | 40% |
| Diffuse large B-cell lymphoma | 31% | 64 | 1.2 : 1 | 25% | 29% | 13% | 33% | 30% | 16% | 19% | 46% | 41% |
| Mediastinal large B–cell lymphoma | 2% | 37 | 0.5 : 1 | 10% | 56% | 3% | 31% | 52% | 3% | 11% | 50% | 48% |
| Burkitt lymphoma | <1% | 31 | 8.1 : 1 | 37% | 25% | 0% | 38% | 22% | 33% | 14% | 44% | 44% |
| Burkitt-like lymphoma | 2% | 57 | 1.4 : 1 | 26% | 26% | 7% | 41% | 42% | 21% | 22% | 47% | 43% |
| Extranodal marginal zone lymphoma, MALT type | 5% | 60 | 0.92 : 1 | 39% | 28% | 2% | 31% | 8% | 14% | 8% | 81% | 65% |
| Marginal zone lymphoma, nodal type | 1% | 58 | 0.7 : 1 | 13% | 13% | 34% | 40% | 0% | 32% | 13% | 56% | 28% |
| Peripheral T-cell lymphoma | 6% | 61 | 1.2 : 1 | 8% | 12% | 15% | 65% | 12% | 36% | 31% | 25% | 18% |
| Anaplastic large cell lymphoma | 2% | 34 | 2.2 : 1 | 19% | 32% | 10% | 39% | 17% | 13% | 21% | 77% | 58% |
B-lymphoblastic leukemia/lymphoma, NOS
B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities
Chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL)
B-cell prolymphocytic leukemia
Splenic marginal zone lymphoma
Hairy cell leukemia
Splenic B-cell lymphoma/leukemia, unclassifiable
Splenic diffuse red pulp small B-cell lymphoma
Hairy cell leukemia variant
Lymphoplasmacytic lymphoma
Heavy chain diseases
Plasma cell myeloma
Plasmacytoma
Solitary plasmacytoma of bone
Extraosseous plasmacytoma
Extranodal marginal zone lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma)
Nodal marginal zone lymphoma
Pediatric nodal marginal zone lymphoma
Follicular lymphoma
In situ follicular neoplasia
Duodenal-type follicular lymphoma
Testicular follicular lymphoma
Pediatric-type follicular lymphoma
Large B-cell lymphoma with IRF4 rearrangement
Primary cutaneous follicle center lymphoma
Mantle cell lymphoma
In situ mantle cell neoplasia
Diffuse large B-cell lymphoma (DLBCL)
DLBCL, NOS
T-cell/histiocyte-rich large B-cell lymphoma
Primary DLBCL of the CNS
Primary cutaneous DLBCL, leg type
EBV+ DLBCL, NOS
EBV+ mucocutaneous ulcer
DLBCL associated with chronic inflammation
Fibrin-associated DLBCL
Lymphomatoid granulomatosis
Primary mediastinal (thymic) large B-cell lymphoma
Intravascular large B-cell lymphoma
ALK+ large B-cell lymphoma
Plasmablastic lymphoma
Primary effusion lymphoma
HHV8-positive DLBCL, NOS
HHV8-positive germinotropic lymphoproliferative disorder
Burkitt lymphoma
Burkitt-like lymphoma with 11q aberration
High-grade B cell lymphoma
High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements
High-grade B-cell lymphoma, NOS
B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classic Hodgkin lymphoma
NOS, Not otherwise specified.
T-lymphoblastic leukemia/lymphoma, NOS
Early T-cell precursor lymphoblastic lymphoma
T-cell prolymphocytic leukemia
T-cell large granular lymphocytic leukemia
Chronic lymphoproliferative disorder of NK cells
Aggressive NK-cell leukemia
EBV+ T-cell and NK-cell lymphoproliferative diseases of childhood
Systemic EBV+ T-cell lymphoma of childhood
Chronic active EBV infection of T-cell and NK-cell type, systemic form
Hydroa vacciniforme–like lymphoproliferative disorder
Severe mosquito bite allergy
Adult T-cell leukemia/lymphoma
Extranodal NK/T-cell lymphoma, nasal-type
Intestinal T-cell lymphoma
Enteropathy-associated T-cell lymphoma
Monomorphic epitheliotropic intestinal T-cell lymphoma
Intestinal T-cell lymphoma, NOS
Indolent T-cell lymphoproliferative disorder of the gastrointestinal tract
Hepatosplenic T-cell lymphoma
Subcutaneous panniculitis-like T-cell lymphoma
Mycosis fungoides
Sezary syndrome
Primary cutaneous CD30+ lymphoproliferative disorders
Lymphomatoid papulosis
Primary cutaneous anaplastic large cell lymphoma
Primary cutaneous peripheral T-cell lymphomas, rare subtypes
Peripheral T-cell lymphoma (PTCL), NOS
Angioimmunoblastic T-cell lymphoma
Follicular T-cell lymphoma
Nodal peripheral T-cell lymphoma with TFH phenotype
Anaplastic large cell lymphoma (ALCL), ALK positive
Anaplastic large cell lymphoma (ALCL), ALK negative
Breast implant–associated anaplastic large cell lymphoma
NOS, Not otherwise specified.
The REAL classification has been validated both in terms of reproducibility and clinical relevance by a large retrospective study on 1378 cases of lymphoma contributed from eight countries. The lymphoma types are confirmed to show distinctive clinical features and behavioral characteristics (see Table 21A.9 ).
Overall there was good intraobserver and interobserver reproducibility in the diagnoses, in the range of 85% or more, which was superior to that of most previous classifications. For many lymphoma types, immunophenotyping contributed significantly to the reproducibility of the diagnoses. Only three lymphoma types had low reproducibility (53%–63%), including Burkitt-like lymphoma, lymphoplasmacytic lymphoma, and nodal marginal zone lymphoma. These were well-recognized problem areas in lymphoma diagnosis, and recent identification of molecular markers (e.g., MYD88 L265P mutation in lymphoplasmacytic lymphoma) can significantly improve diagnostic accuracy.
Classification of NHLs is important for patient management and prognostication because each lymphoma type exhibits distinctive clinicopathologic features. As a generalization, there are three major categories of lymphoma in terms of biologic behavior: indolent lymphoma, aggressive lymphoma, and highly aggressive lymphoma—so-called the good, the bad, and the ugly, respectively ( Table 21A.10 ). It is somewhat paradoxic that, in the long term, the survival of aggressive and highly aggressive lymphomas is better than that of indolent lymphomas because of curability of the disease. Two lymphoma types exhibit features distinct from the abovementioned three categories, and they include primary cutaneous ALCL and extranodal marginal zone lymphoma of MALT type, both of which have a favorable prognosis and may regress spontaneously or after eradication of the antigenic source. This simplified overview can serve as a conceptual framework to help in understanding and learning the various lymphoma types.
As shown in Table 21A.9 , the two commonest NHL types in adults are DLBCL and follicular lymphoma. Together they account for more than 50% of all cases. T–cell lymphomas are relatively uncommon.
In children, aggressive lymphomas predominate, with lymphoblastic lymphoma (~35%), Burkitt lymphoma (~40%), and large cell lymphoma (~20%) accounting for almost all cases. The large cell lymphoma category includes predominantly ALCL and DLBCL.
The International Prognostic Index (IPI), which incorporates several parameters, is extremely useful in stratifying patients into different prognostic groups for each lymphoma type ( Box 21A.5 ). It is applicable for both indolent and aggressive lymphomas. Some studies have made modifications for individual lymphoma types. Examples are follicular lymphoma IPI, mantle cell lymphoma IPI, revised IPI for diffuse large B-cell lymphoma, and prognostic index for peripheral T-cell lymphoma unspecified.
Age: ≥60 years
Stage: Advanced stage (III or IV)
Number of extranodal sites of involvement: >1
Performance status: ≥2
Serum LDH level: Abnormal
Total number of above features that are present.
| 0–1: | Low risk |
| 2: | Low intermediate risk |
| 3: | High intermediate risk |
| 4–5: | High risk |
Little is known about the etiology of NHLs. The wide variations in the incidences of some lymphoma types in different ethnic populations suggest genetic predisposition and environmental factors as being contributory (e.g., follicular lymphoma, chronic lymphocytic leukemia/small lymphocytic lymphoma, and enteropathy-associated T-cell lymphoma are much less common in Asian and developing countries compared with Western countries, while extanodal NK/T-cell lymphomas are more prevalent in Asian populations). Genome-wide association studies have already identified some genetic loci that increase the susceptibility for CLL/SLL and extranodal NK/T-cell lymphoma. Some indolent lymphomas, most notably CLL/SLL, show a clear association with advancing age, suggesting an etiologic relationship with decreasing diversity of the immune repertoire.
Immunodeficiency (congenital, acquired, or iatrogenic) and autoimmune diseases predispose to the development of malignant lymphomas. The defective immune surveillance permits the emergence of abnormal clones with proliferative advantage, such as EBV-infected cells. Chronic irritation or suppuration in a confined space may also provide a microenvironment for the emergence of lymphoma.
Some lymphoma types have been associated with infective agents, including bacteria and viruses, as detailed in Table 21A.11 . The importance of recognition of the association is several-fold: (1) demonstration of EBV or human herpesvirus-8 (HHV8) can aid in the diagnosis of some lymphoma types; (2) eradication of the infective agent can lead to regression of gastric extranodal marginal zone lymphoma, immunoproliferative disease of the small bowel, and hepatitis C–associated lymphoma (some cases); (3) restoration of immune competence can lead to regression of some EBV–associated lymphoproliferative disorders; (4) some virus-associated lymphomas can be treated by immune modulators (e.g., lymphomatoid granulomatosis, adult T-cell lymphoma/leukemia); and (5) preventive measures or vaccination may potentially reduce the incidence of some lymphoma types associated with infective agents.
| Infective Agent | NHL Types Associated With the Infective Agent | Treatment Implications and Remarks |
|---|---|---|
| Helicobacter pylori |
|
Eradication of Helicobacter results in regression of the lymphoma in >70% of cases. |
| Campylobacter jejuni |
|
Regression can occur with tetracycline therapy. |
| Epstein-Barr virus (EBV) |
|
Reduction in immunosuppressant can lead to regression of some posttransplant lymphoproliferative disorders and methotrexate-associated lymphoproliferative disorders. Lymphomatoid granulomatosis may respond to immunologic manipulation (e.g., interferon-α). EBV+ mucocutaneous ulcer is self-limiting or can resolve with reduction in immunosuppression. |
| Human herpesvirus 8 (HHV8) |
|
Homologues of some human cellular genes are encoded by HHV8 |
| Human T-lymphotropic virus I |
|
Endemic in Japan, northern Taiwan, Brazil, and Caribbean area. The disease sometimes responds to antiviral therapy (zidovudine plus interferon-α) |
| Hepatitis C virus |
|
There are geographic variations in the association of hepatitis C virus with B-cell lymphomas, and the association is strongest in Italy and Japan. The virus is not integrated into the lymphoma cells. It is postulated that the neoplastic clone evolves from the background of hepatitis C antigen-driven chronic B-cell stimulation; hepatitis C infection also induces a mutator phenotype in B cells. Up to 75% of cases of low-grade B-cell lymphomas associated with hepatitis C regress when treated with antiviral agents and/or interferon. |
| Hepatitis B virus |
|
Hepatitis B-associated DLBCLs are diagnosed at a younger age, with high IPI, more advanced stage disease, and a worse prognosis. The lymphoma does not arise through chronic antigenic stimulation, but appears to result from genetic alterations resulting from chronic infection. The lymphoma does not respond to antiviral therapy. |
| Chlamydia psittaci |
|
The association of C. psittaci infection with ocular adnexal extranodal marginal zone lymphoma is controversial. A strong association has been reported in Italy, Germany, and some other countries, but some studies fail to document such an association. There have been reports of regression of ocular adnexal extranodal marginal zone lymphoma after treatment with antibiotics for C. psittaci . |
| Borrelia burgdorferi |
|
The association of B. burgdorferi with cutaneous B-cell lymphomas is seen almost exclusively in the United Kingdom, with some cases regressing after B. burgdorferi eradication therapy. |
Although a few lymphoma types can be diagnosed reliably by morphologic assessment alone (such as chronic lymphocytic leukemia/small lymphocytic lymphoma, follicular lymphoma, and extranodal marginal zone lymphoma of MALT), immunophenotypic analysis constitutes an integral workup for lymphomas because it improves the accuracy of classification and provides important information for targeted therapy (such as rituximab for CD20-expressing lymphomas). In the descriptions detailing the various lymphoma types, the markers will be listed by the following convention:
Markers indicating the lineage of the lymphoma
Markers that are helpful in defining the entity or helpful for diagnosis
Documented immunophenotype, but generally without diagnostic significance
Marker profile that can potentially lead to an erroneous diagnosis
The immunophenotypic profile of a lymphoma may change after therapy (e.g., treatment with anti-CD20 antibody [rituximab] may result in loss of CD20 expression by the residual or relapsed lymphoma cells). In such situations, use of other B–lineage markers (such as PAX5) is required to document their cellular lineage.
Some cases of lymphoma (a few percent) defy classification even after exhaustive workup. They can be labeled malignant lymphoma, unclassifiable . If possible, provide the information on the lineage and a best guess as to whether the tumor is low grade or high grade. The reason for the difficulties in classification should be stated, such as failure to fit into the defined categories of lymphoma, insufficient tissue sample, suboptimal tissue preservation, unavailability of special studies, and unavailability of clinical information. If the reason is inadequate tissue sample, the problem can often be solved by securing further material for examination.
Lymphoblastic lymphoma is a highly aggressive neoplasm composed of precursor cells of T, B, or rarely NK cell lineage. Lymphoblastic lymphoma and acute lymphoblastic leukemia represent different ends of the spectrum of the same biologic entity. In most studies, an arbitrarily adopted criterion of less than 25% lymphoblasts in marrow is used to distinguish the former from the latter. Overall, 80% to 90% of all lymphoblastic lymphomas are of T-cell lineage. For B–cell lymphoblastic neoplasms, most cases present as acute lymphoblastic leukemia, while a lymphomatous presentation is rare.
T-lymphoblastic lymphoma most commonly affects adolescents and young adults, but no age group is exempt. The male to female ratio is 2 : 1. Most patients present with respiratory difficulties due to a large mediastinal mass, often accompanied by symptoms of superior vena cava obstruction, pleural effusion, and supraclavicular lymphadenopathy. Other patients present with lymphadenopathy or extranodal disease. More than 85% of patients have advanced disease (stage III or IV) at presentation. There is a tendency of early dissemination to the bone marrow and central nervous system.
A prompt diagnosis of lymphoblastic lymphoma is important so that treatment can be initiated without delay for this rapidly growing tumor. Although the prognosis was previously dismal, this lymphoma is now highly curable using aggressive chemotherapy similar to that used for acute lymphoblastic leukemia, with survival rates of 80% to 90% being achieved in children and around 60% in adults.
Rare cases of lymphoblastic lymphoma associated with tissue and peripheral blood eosinophilia have been reported to develop acute myeloid leukemia, myelodysplastic syndrome, or myeloid hyperplasia synchronously and subsequently. This highly aggressive syndrome is termed myeloid/lymphoid neoplasms with eosinophilia and rearrangement of PDGFRA, PDGFRB, or FGFR1, or with PCM1-JAK .
Exceptional examples of indolent T-lymphoblastic proliferation pursuing a protracted course with surgical excision alone have also been reported. The histologic and immunophenotypic features are practically indistinguishable from T–lymphoblastic lymphoma. The only differences are the presence of a spectrum of cells similar to the immature lymphoid cells seen in the normal thymic cortex and the absence of clonal TR gene rearrangements.
B-lymphoblastic lymphoma occurs over a wide age range, from children to older adults, with a mean age of 32 years. There is slight female predominance. The patients present with prominent lymphadenopathy or extranodal disease, the predilection sites being the skin and bone. The skin lesions occur most commonly in the head and neck region, and the bone lesions are lytic and involve a single or multiple sites. Mediastinal involvement is rare. The overall survival is better than that of T-lymphoblastic lymphoma, at least in the initial 2 to 3 years. The prognosis of localized disease is particularly favorable.
This entity is so rare that little is known about its clinical and behavioral features. Assessment of its clinical behavior and prognosis is complicated by the fact that many cases previously diagnosed as NK-lymphoblastic leukemia/lymphoma, based on CD56 expression, are now recognized as blastic plasmacytoid dendritic cell neoplasms.
The lineage cannot be predicted from the histologic findings. The key morphologic feature of lymphoblastic lymphoma/leukemia is a diffuse, dense, permeative, monomorphous infiltrate of midsize lymphoid cells with thin nuclear membranes, delicate chromatin, indistinct nucleoli, and a high mitotic count. The nuclei can be round or convoluted; the convoluted nuclei show multiple deep foldings while still maintaining an overall rounded contour ( Figs. 21A.36 and 21A.37 ). The cytoplasm is typically scanty, giving an impression of nuclear crowding. There is little variation in the size of the cells, and multinucleated tumor cells are rare. There may be interspersed tingible-body macrophages, imparting a starry-sky appearance.


Effacement of the nodal architecture can be complete or partial, and infiltration of the perinodal tissue often produces a single-file pattern ( Fig. 21A.38 ). The nuclei may appear elongated when the lymphoma cells infiltrate fibrous tissue. Occasionally, stretching of the reticulin framework or blood vessels results in a multinodular (pseudofollicular) pattern that may lead to a misdiagnosis of follicular lymphoma ( Fig. 21A.39 ).


Terminal deoxynucleotidyl transferase (TdT) is the defining marker for lymphoblastic neoplasms and helps in distinction from peripheral T-cell lymphomas or mature B–cell lymphomas. TdT immunostaining is localized to the nuclei ( Fig. 21A.40 ). CD99, an antigen typically expressed in Ewing sarcoma, is an alternative but less specific marker for precursor cell (lymphoblastic) neoplasms.
 The neoplastic cells show positive staining for CD3. (Right) Nuclear staining for TdT is evident.")
Lineage assignment can be problematic in lymphoblastic lymphomas, because CD20 may be negative in B–lymphoblastic lymphoma, CD3 may be negative in T-lymphoblastic lymphoma, and CD79a immunoreactivity can occur in some T–lymphoblastic lymphomas. Rare cases are biphenotypic (expressing both T and B markers, or coexpressing myeloid markers) or show a null-cell immunophenotype.
CD7, which is expressed from a very early stage of T-cell development, is most consistently expressed. The pan-T markers CD2, CD3 (cytoplasmic CD3+, surface CD3+/-), and CD5 are variably positive (see Fig. 21A.40 ).
TdT and CD99 are positive (see Fig. 21A.40 ). CD1a and CD10 are variably expressed. CD43 is often but not always positive, while CD45RO is frequently negative. TAL1 is expressed in 50% to 75% of cases, independent of whether there is structural alteration of the TAL1 gene. LMO2, a marker commonly expressed in normal germinal center cells and follicular lymphomas, is positive in T-lymphoblastic lymphomas, and its expression can aid in distinction from normal thymic tissue or thymoma (LMO2 negative) in small biopsy samples.
The variable expression of CD4 and CD8 results in a CD4+ CD8+, CD4- CD8-, CD4+ CD8-, or CD4- CD8+ phenotype. TCR may be of αβ or γδ type, or may not be expressed. The rare cases expressing NK-cell–associated markers, such as CD16 or CD57, have been found to pursue a more aggressive course.
In 10% of T–lymphoblastic lymphomas, CD79a is coexpressed with CD3, causing problems in lineage determination.
Approximately 10% of cases of T-lymphoblastic leukemia/lymphoma, and 5% to 10% in adults, have an immunophenotype indicative of very early and limited T-cell differentiation, including lack of expression of CD8 and CD1a and expression of one or more stem cell/myeloid markers (CD34, CD117, HLA-DR, CD33, CD11b). These cases, which may be difficult to distinguish from acute myeloid leukemia, are termed early T-cell precursor lymphoblastic leukemia .
B-cell marker positive (e.g., CD19, PAX5, and CD79a). PAX5 and CD19 are most consistently expressed. CD20 is not uncommonly negative. Cytoplasmic Ig µ-chain can be demonstrated in some cases, but surface Ig expression is uncommon.
TdT+, and often CD10+. CD99 is usually positive.
HLA-DR+, CD34+/-.
Some cases may show punctate staining for cytokeratin. CD43 is frequently positive.
Surface CD3-, cytoplasmic CD3+, CD5-, CD56+.
TdT+.
T-lymphoblastic lymphomas show clonal rearrangement of TRB and TRG genes in approximately 95% of cases, but about 20% may in addition show rearrangement of IGH but not IGK and IGL genes. Numerous cytogenetic abnormalities have been described, and many of the translocations implicate the TR genes on 14q11 ( TRA and TRD ) or 7q35 ( TRB ). Many partner genes have been involved in the translocations (e.g., TAL1 [1p32-34], TTG2/RHOM2 [11p13], HOX11 [10q24], and MYC [8q24]). TAL1 is the most commonly involved gene, in the form t(1; 14)(p32-34; q11) or, more commonly, 90–kb interstitial deletion. The latter results in juxtaposition of TAL1 with an upstream locus SIL , resulting in overexpression of TAL1 as TAL1 is brought under the control of the regulatory elements of SIL .
Approximately 50% of cases harbor activating mutations in the gene encoding the lineage-directing transcription factor NOTCH1 . An additional subset harbors inactivating mutations in the gene encoding FBXW7 , a negative regulator of NOTCH1 signaling. Persistent NOTCH1 activity is required for the growth of T-ALL cell lines in vitro, through induction of MYC. Whole genome and targeted sequencing of pediatric cases has revealed recurrent mutations in genes regulating cytokine receptor signaling and RAS signaling, genes governing hematopoietic development, and genes encoding histone modifying proteins in a proportion of cases.
Gene expression profiling studies have identified several signatures indicative of leukemic arrest at specific stages of normal thymocyte development: LYL1+ signature (pro-T), HOX11+ (early cortical thymocyte), and TAL1+ (late cortical thymocyte). HOX11 activation is significantly associated with a favorable prognosis, while expression of TAL1 or LYL1 is associated with a much worse response to treatment.
B-lymphoblastic lymphoma shows clonal rearrangement of IGH genes in approximately 100% of cases, but 20% to 30% can show concurrent clonal rearrangement of TRB genes. Many different cytogenetic aberrations have been recognized in acute lymphoblastic leukemia of B lineage, with t(9; 22)(q34; q11), t(1; 19)(q23; p13), t(4; 11)(q21; q23), and hypodiploidy being associated with an unfavorable prognosis and hyperdiploidy with more than 50 chromosomes being associated with a favorable prognosis. Chromosomal rearrangements resulting in deregulated expression of the cytokine receptor CRLF2 occurs in approximately 5% of cases. Sequencing studies have revealed recurrent genetic mutations in PAX5, IKZF1, JAK2 , and CRLF2, which are thought to arrest normal B-cell development ( PAX5, IKZF1 ) and promote cytokine-mediated proliferation ( JAK2 , CRLF2 ). Little is known about the pattern or the significance of cytogenetic abnormalities in the lymphomatous form of the disease.
Myeloid sarcoma. Myeloid sarcoma can closely mimic lymphoblastic lymphoma. It is always prudent to exclude this possibility, particularly if any of the following suspicious features are present:
Nucleoli more prominent than usual
Many nuclei with reniform contour
Distinct nucleoli
Interspersed eosinophilic myelocytes
Fine granularity in the cytoplasm
CLL/SLL. In suboptimally fixed materials, lymphoblasts may appear shrunken and dark, mimicking small lymphocytes. The young age of the patient and the brisk mitotic activity should alert one to the possibility of lymphoblastic lymphoma. Examination of histologic sections with light hematoxylin stain (such as periodic acid–Schiff [PAS] or immunostained slide) is helpful in appreciating the delicate chromatin pattern and mitotic figures of lymphoblasts.
Normal thymic tissue and thymoma (see Chapter 21C ).
Indolent T-lymphoblastic proliferation.
Mantle cell lymphoma, blastoid variant. Lymphoblastic lymphoma is TdT+ cyclin D1–, while mantle cell lymphoma shows the opposite pattern of immunoreactivity.
Follicular lymphoma. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” later in the chapter.
Burkitt lymphoma ( Table 21A.12 ). See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” later in the chapter.
| Lymphoblastic Lymphoma | Burkitt Lymphoma | |
|---|---|---|
| Nuclei | Round or convoluted; usually no significant molding | Usually round, but occasionally may show nuclear protrusions; prominent nuclear molding, with squaring of nuclear membrane |
| Chromatin pattern | Fine, dusty | Coarsely granular |
| Nucleoli | Inconspicuous | Distinct, two to five nucleoli, often not membrane bound |
| Cytoplasm | Scanty and barely visible in routine histologic sections. In Giemsa-stained touch preparations, cytoplasm is apparently found only around part of the perimeter of the nucleus. | Definite rim of basophilic to plasmacytoid cytoplasm, with squaring of cytoplasmic outline. In Giemsa-stained touch preparations, small lipid vacuoles are found in the basophilic cytoplasm. |
| Lineage | Usually T lineage, sometimes B or NK lineage | Always B lineage |
| Ki67 index | High, but usually <80% | ~100% |
Blastic plasmacytoid dendritic cell neoplasm.
Mature B-cell lymphomas include neoplasms of virgin/naïve B cells, follicular/germinal center cells, postgerminal center cells, and memory B cells that have undergone extrafollicular T-cell–independent antigen response. These different stages of B-cell differentiation can be defined by the mutation pattern of the rearranged IG genes as well as immunohistochemical markers. Follicular center cells typically show somatic hypermutations of the IG genes, with evidence of intraclonal diversity (ongoing mutation). Somatic hypermutation refers to the presence of mutations, mainly in the form of single nucleotide exchange, but sometimes deletions or duplications introduced at a high rate into the variable region genes. This is a mechanism for generating additional diversity in the IG gene so as to produce high-affinity Ig molecules.
Chronic lymphocytic leukemia/small lymphocytic lymphoma is a neoplastic proliferation of nonactivated, mature-looking small lymphocytes. Most cases have a leukemic presentation (CLL), and only rare cases have localized disease (SLL); they represent different manifestations of the same biologic entity. If there is documented lymphocytosis at the time of diagnosis, CLL can be diagnosed. If such information is not known, a diagnostic label CLL/SLL is given.
CLL/SLL typically occurs in older subjects (see Table 21A.9 ). Most patients are incidentally found to have lymphocytosis, lymphadenopathy, or splenomegaly. Lymphadenopathy, when present, is often generalized, and bone marrow involvement is common. About 40% of patients have B symptoms, which predict a shorter survival. Paraproteinemia is uncommon. Patients are prone to develop infection complications, and some may develop paraneoplastic phenomena, including autoimmune hemolytic anemia and pure red cell aplasia, the latter due to production of antibodies against the erythropoietin receptor as seen in association with thymoma. For CLL, the Rai or Binet staging system is usually used.
CLL/SLL is indolent, and asymptomatic patients can be observed without specific treatment. For patients requiring treatment, there is good response to chemoimmunotherapy (FCR: fludarabine, cyclophosphamide, and rituximab; or BR: bendamustine and rituximab), sometimes with complete remission. Newer therapies, such as ibrutinib (Bruton tyrosine kinase inhibitor), idelalisib (PI3Kδ inhibitor), and venetoclax (BCL2 inhibitor), used alone or in combination with chemoimmunotherapy, are also highly effective for relapsed or high-risk disease. The clinical course is characterized by repeated relapses and eventually death after many years. Although the median survival is approximately 10 years, this average conceals significant heterogeneity in prognosis. For patients with Binet stage A disease at diagnosis and mutated IG genes, median survival exceeds 20 years. However, there is a subgroup of patients whose disease is rapidly progressive. Features associated with a poor prognosis include high clinical stage, high white cell count (>50 × 10 9 /L), high percentage of circulating prolymphocytes (>10%), cytogenetic abnormality of del(11q) or del(17p), CD38 expression, ZAP70 expression, and unmutated IG genes.
The lymph node shows diffuse effacement of architecture, although in exceptional cases the neoplasm selectively involves the interfollicular regions or B zones of the node. The infiltrate commonly spills over into the perinodal tissue. In most cases, the dark-staining lymphoid infiltrate is characteristically interspersed with pale round foci of proliferation centers (pseudofollicles), and this pattern alone is pathognomonic of CLL/SLL ( Fig. 21A.41 ). Nonetheless, proliferation centers are not present in some cases.

The lymphoma cells are small, with round nuclei, condensed chromatin, inconspicuous nucleoli, and scanty cytoplasm. A mild or moderate degree of nuclear irregularity can sometimes be observed and may potentially lead to an erroneous diagnosis of mantle cell lymphoma. Plasma cells are sparse or absent. The mitotic count is usually low. The proliferation centers are typically nonexpansile and comprise a mixture of prolymphocytes and paraimmunoblasts ( Figs. 21A.42 and 21A.43 ). Prolymphocytes are slightly bigger than small lymphocytes and have more open chromatin, a distinct nucleolus, and a greater amount of pale cytoplasm. Paraimmunoblasts are even larger, with vesicular chromatin and a prominent central nucleolus; they can be distinguished from immunoblasts by being slightly smaller and having paler cytoplasm. Besides being present in proliferation centers, prolymphocytes and paraimmunoblasts are also interspersed individually among small lymphocytes (see Figs. 21A.42A and 21A.43 ). They are the hallmark of CLL/SLL and permit this diagnosis to be made with confidence even if their nuclei or the nuclei of the small cells appear irregular (see Fig. 21A.42B ). Exceptionally, pseudorosettes can be formed.
 Paraimmunoblasts and prolymphocytes are typically intimately intermingled with the major population of small lymphocytes, which show round nuclear contour, condensed chromatin, and scanty cytoplasm. (B) Rare cases can show marked nuclear lobations or foldings.")

Cases showing expanded proliferation centers (broader than 20× field) or high proliferative rate (>40%/proliferation center) are considered to have accelerated CLL. In patients with atypical CLL (defined as presence of >10% prolymphocytoid cells or >10% cells with cleaved nuclei or plasmacytoid features), the lymph nodes show much larger proliferation centers, and the lymphoid cells often exhibit nuclear irregularities. The term paraimmunoblastic variant is applied when there are sheets or large aggregates of paraimmunoblasts; this variant is associated with a worse prognosis ( Fig. 21A.44 ).

In some cases, lymphoplasmacytoid cells are present in an otherwise histologically typical CLL/SLL. These cells have the nuclear features of small lymphocytes but possess a moderate rim of amphophilic cytoplasm that lacks a distinct Golgi zone ( Fig. 21A.45 ). Plasma cells are, however, rare. Cytoplasmic Ig can be demonstrated, and there may be paraproteinemia. This subgroup corresponds to lymphoplasmacytoid immunocytoma in the Kiel classification.

Pan-B markers (e.g., CD19, CD20, CD79a) positive; however, CD20 expression is often weak. Monotypic surface Ig (usually both IgD and IgM) expression is also weak.
Usually CD5+, CD10-, CD23+, LEF1+, CD200+ ( Fig. 21A.46 ). Cyclin D1 is usually negative, but some cells in proliferation centers can be positive; the cyclin D1 positive cells do not show SOX11 immunoreactivity, and they lack CCND1 translocation.
 CD20 is positive, but is often weak as shown here. (Middle) The neoplastic cells express CD5. (Right) CD23 is commonly expressed.")
Ki67 (proliferation) index is usually low except in proliferation centers. A Ki67 proliferation index above 25% predicts a less favorable outcome. TCL1 is commonly expressed. Reactive T cells are generally sparse in the background.
Since CD20 expression can be very weak or absent while CD43 is commonly positive, immunophenotyping using CD20 and CD43 alone (i.e., without another pan-B-cell marker) can potentially lead to misinterpretation as T-cell lymphoma.
IG genes are clonally rearranged. Although previous studies suggest that CLL/SLL is a naïve B-cell neoplasm with unmutated IG gene, recent studies have clearly shown that this disease is derived from antigen-experienced B lymphocytes that differ in the level of IG V-gene mutations. It has been postulated that CLL/SLL may be derived from marginal zone B cells. Approximately 50% of the cases show unmutated IG genes, and approximately 50% show somatic hypermutations. The unmutated group is associated with a worse prognosis, with a median survival of 95 months compared to 293 months in the mutated group. Gene expression profiling studies of CLL/SLL have shown a homogeneous phenotype most resembling memory B cells, and the unmutated and mutated groups exhibit differential expression of a restricted number of genes, with ZAP70 gene overexpression best distinguishing the unmutated from the mutated group. Immunohistochemical expression of ZAP70 can serve as a surrogate marker for the unmutated status.
At diagnosis, CLL frequently shows deletions of 13q14 (~50% of cases), which can be homozygous or hemizygous. Two micro-RNA genes located at this chromosome region are deleted or downregulated in most cases of CLL. Deletion in 11q22-q23, found in approximately 14% of CLL/SLL, is associated with extensive lymph node involvement and poorer survival. The implicated gene is ATM (mutated in ataxia-telangiectasia), and somatic disruption of both alleles by deletion or point mutation appears to play a pathogenic role in CLL/SLL. Deletions of 17p13 and mutation/deletion of TP53 gene occur in about 10% of CLL and are correlated with late stages of the disease and poor survival. Trisomy 12 is found in about 15% of CLL but is observed in only subpopulations of tumor cells and thus probably reflects progression rather than a primary genetic alteration.
Whole exome sequencing of CLL has confirmed the importance of mutations in the tumor-suppressor molecules TP53 and ATM and also highlighted recurrent mutations in genes encoding the signaling molecules MYD88 (10% of cases), active in NFκB signaling, and NOTCH1 (4% of cases), active in determining cell fate. Mutations in NOTCH1 are typically dinucleotide deletions in the PEST domain, which result in decreased ubiquitination and increased NOTCH1 activity; the presence of such mutations is associated with decreased overall survival. Approximately 15% of cases harbor mutations in SF3B1 , which regulates pre-mRNA splicing and is associated with poor clinical outcome.
In a proportion of CLL/SLL (3%–10%), a large B-cell lymphoma can supervene (Richter syndrome), when the disease will pursue an accelerated downhill course, eventuating in death within 1 year. The large B-cell lymphoma is composed of large cells, which frequently show significant cellular pleomorphism and bizarre forms, including Reed-Sternberg–like cells ( Fig. 21A.47 ). Molecular studies have shown that the large cell lymphoma is either clonally related (true transformation) or clonally unrelated to the CLL (a new neoplastic event in a predisposed host). Rarely, other types of aggressive lymphoma can also supervene.
. The chronic lymphocytic leukemia component is seen in the left field. The large cell lymphoma component is seen in the right field. Large pleomorphic and Reed-Sternberg–like cells are common (inset).")
On rare occasions, isolated large cells resembling Reed-Sternberg cells are dispersed among the small lymphoma cells, but without the inflammatory background typical of CHL ( Fig. 21A.48 ). These Reed-Sternberg–like cells can show an immunophenotype of activated B cells, typical Reed-Sternberg cells, or a hybrid immunophenotype of B cells and Reed-Sternberg cells. EBV plays a key role in the genesis of these large cells since EBV has been consistently demonstrated in them but not in the CLL/SLL cells.
, there are singly scattered Reed-Sternberg cells (shown to CD20+, CD30+). This does not qualify for a diagnosis of Hodgkin lymphoma because the inflammatory background (such as T cells) required for this diagnosis is lacking.")
Sometimes CHL, defined by the presence of RS cells accompanied by an appropriate inflammatory background, can supervene. This phenomenon is seen at greater frequency in recent years, related to increased use of fludarabine (which is highly immunosuppressive) for treatment of CLL/SLL. The CLL/SLL and HL may or may not be clonally related.
NLPHL. Particularly in young patients, NLPHL must be excluded before a diagnosis of CLL/SLL is rendered. Immunohistochemically, the small lymphocytes of CLL/SLL are monotypic and aberrantly express CD5 and LEF1, while those of NLPHL are polytypic and do not exhibit an aberrant immunophenotype.
Other small B-cell lymphomas.
Lymphoblastic lymphoma.
Reactive lymphoid hyperplasia.
Follicular lymphoma. The prominent proliferation centers seen in CLL/SLL may invite an erroneous interpretation of follicular lymphoma. In contrast to neoplastic follicles, the proliferation centers are nonexpansile and are usually composed of cells with round nuclei and central nucleoli (prolymphocytes and paraimmunoblasts). Reticulin stain will highlight condensed fibers around neoplastic follicles of follicular lymphoma but not around proliferation centers. CLL/SLL is CD5+ CD10-, while follicular lymphoma is often CD5- CD10+.
See Chapter 22 .
Lymphoplasmacytic lymphoma (LPL) is an uncommon low-grade B-cell lymphoma composed of small lymphoid cells with variable degrees of plasmacytic differentiation. It corresponds to lymphoplasmacytic (not lymphoplasmacytoid) immunocytoma in the Kiel classification. Until recently, it was a diagnosis of exclusion; however, identification of the causal MYD88 L265P mutation has greatly facilitated its diagnosis. Although Waldenstrom macroglobulinemia (defined as LPL with bone marrow involvement and an IgM monoclonal gammopathy of any concentration) is common, it is not essential for diagnosis.
LPL most commonly occurs in middle-aged and elderly people and is usually disseminated at presentation (see Table 21A.9 ). The patients present with lymphadenopathy, splenomegaly, or symptoms of hyperviscosity syndrome (Waldenstrom macroglobulinemia), with fatigue, headache, and visual disturbance. Involvement of the central nervous system, known as Bing-Neel syndrome, is a rare but feared complication.
Lymphocytosis is sometimes seen, and the marrow is frequently involved. Paraproteinemia is a common finding; it is usually of IgM, but sometimes IgG or IgA type. Some patients may have Coombs-positive hemolytic anemia. The disease pursues an indolent relapsing course. The median survival is 6.5 to 9 years. A recent study reports superior response to ibrutinib-rituximab therapy. Some cases can transform to a large B–cell lymphoma (EBV usually negative), when the disease will pursue a rapidly downhill course.
The nodal architecture may or may not be obliterated, but the sinuses are often preserved or distended with proteinaceous fluid ( Fig. 21A.49 ). The pattern is often diffuse, and proliferation centers are absent. Small lymphocytes are admixed with lymphoplasmacytoid cells and mature-looking plasma cells. Dutcher bodies, which are brightly eosinophilic, PAS–positive globules in the nuclei, are commonly present, and they represent invaginations of cytoplasmic Ig into the nuclei ( Fig. 21A.50 ). Globular inclusions (Russell bodies) or crystalline inclusions of Ig are not uncommonly seen in the cytoplasm of the lymphoplasmacytoid cells and plasma cells ( Fig. 21A.51 ). Some large activated lymphoid cells may be admixed. Mast cells are often increased. In some cases, there may be an extensive epithelioid histiocytic reaction. There can be extracellular deposits of Ig in the form of amorphous masses or crystals, sometimes accompanied by foreign body giant cell reaction. Rare cases are associated with numerous crystal-storing histiocytes, mimicking adult rhabdomyoma.
.")
 are prominent.")

The morphologic spectrum of LPL has broadened with the availability of MYD88 mutation as a diagnostic aid. In some cases, the neoplastic cells comprise almost exclusively small lymphocytes, although cytoplasmic immunoglobulin can be demonstrated on immunostaining. In other cases, the neoplastic cells comprise almost exclusively plasma cells, mimicking plasmacytoma. Some examples have an appreciable number of admixed large cells, representing disease progression ( Fig. 21A.52 ).

B-lineage markers (e.g., CD20, CD79a) positive. Monotypic cytoplasmic Ig, usually of IgM type, rarely of IgG or IgA type. Surface Ig can often be demonstrated as well ( Fig. 21A.53 ).
 there are few cells staining for κ light chain (residual plasma cells). (Right) Numerous cells stain for λ light chain.")
CD5, CD10, and CD23 are usually negative, but rare cases can be CD5 or CD10 positive. Expression of CD23 is also not uncommon according to some studies. The neoplastic plasma cells are CD138 positive, but in contrast to normal plasma cells they may express CD19 and PAX5.
IG genes are clonally rearranged. Whole genome sequencing has revealed that greater than 90% of cases of LPL with Waldenstrom macroglobulinemia harbor a specific point mutation in the MYD88 gene that results in an amino acid change (L265P) in the encoded protein, which enhances MYD88-mediated NFκB signaling. This mutation is not completely specific for LPL, as it is also found in 30% of cases of nongerminal center–type diffuse large B-cell lymphoma and in rare cases of CLL and marginal zone lymphoma; however, it is absent in myeloma. In addition, mutations in the CXCR4 gene are identified in approximately 30% of cases, nearly always co-occurring with MYD88 mutation; the presence of this mutation is associated with more aggressive clinical behavior and resistance to ibrutinib therapy.
Other small B-cell lymphomas. In particular, distinction from marginal zone lymphoma is most difficult because both LPL and marginal zone lymphoma lack positive defining immunohistochemical markers. In general, a diagnosis of LPL should rarely be made in a mucosal site (for which a diagnosis of extranodal marginal zone lymphoma is more likely). Molecular studies for MYD88 mutation are frequently necessary.
Peripheral T-cell lymphoma with lymphoepithelioid pattern (Lennert lymphoma). Lymphoplasmacytic lymphoma can be recognized by the presence of many plasma cells and plasmacytoid cells. The distinction is more readily made by immunohistochemical studies.
Plasmacytoma. Plasmacytoma is composed of a monomorphous population of plasma cells instead of a mixed population of lymphocytes, plasma cells, and lymphoplasmacytoid cells.
Mantle cell lymphoma is a B-cell lymphoma of mantle zone or primary follicle lymphocytes. It corresponds to centrocytic lymphoma in the Kiel classification. Cyclin D1 overexpression is a hallmark of this neoplasm, although a rare cyclin D1 negative variant is also recognized.
Mantle cell lymphoma typically occurs in middle-aged or older adults. It shows striking male predominance, with the M:F ratio (up to 7 : 1) being the highest among the commoner lymphoma types. The patients often present with lymphadenopathy, but extranodal involvement is also common, especially of the spleen and gastrointestinal tract. Gastrointestinal tract involvement may or may not produce symptoms; some patients present with myriads of polyps in the ileocecal region (lymphomatous polyposis). Most patients have high stage disease, and marrow involvement is common (see Table 21A.9 ).
The clinical course is progressive. There is a poor response to chemotherapy (remission rate only ~50%) ; most responding patients relapse within 20 months. Although the addition of rituximab to combination chemotherapy produces a higher overall response rate (>90%), this has not translated into a prolonged progression-free survival. The median overall survival is 3.5 years, and the 5-year overall survival rate is only about 30%. In more recent studies, the addition of high-dose Ara-C to an R-CHOP–like regime followed by myeloablative consolidation has achieved a significant improvement in progression-free survival. Emerging strategies include ibrutinib (Bruton tyrosine kinase inhibitor), lenalidomide (immune modulatory drug), bortezomib (proteasome inhibitor), and temsirolimus (mTOR inhibitor). Mantle cell lymphoma may show transformation to the blastoid form, but transformation to a DLBCL is extremely rare.
A small subset of patients with mantle cell lymphoma are asymptomatic and have indolent disease that requires little or no treatment. These patients do not have B symptoms or extensive nodal disease characteristic of typical MCL at presentation, but rather show peripheral lymphocytosis with or without splenic involvement. The neoplastic cells tend to be small with clumped chromatin, mimicking small lymphocytes. While cyclin D1 is usually positive, SOX11 is often negative.
The lymphoid infiltrate is diffuse, vaguely nodular, or nodular. Sometimes the neoplastic cells proliferate as broad mantles around residual reactive germinal centers, with coalescence and extension into the interfollicular regions ( Fig. 21A.54 ). Proliferation centers should be absent. Large transformed cells are absent or present only focally in small numbers, probably representing residual lymphoid cells. There are typically interspersed naked pale nuclei representing FDCs ( Fig. 21A.55 ). Scattered solitary epithelioid histiocytes are commonly present and constitute a helpful diagnostic clue ( Fig. 21A.56 ). The blood vessels often show hyalinization, and thick reticulin fibers are commonly present.
 Diffuse growth pattern. This case shows the classic cytology of mantle cell lymphoma, with irregular nuclei, dark chromatin, and scanty cytoplasm. (B) Nodular growth pattern, which may lead to a misdiagnosis of follicular lymphoma. (C) Mantle zone growth pattern around residual germinal centers.")
 Classic type. The small lymphoid cells show nuclear foldings and fairly condensed chromatin. There are scattered larger naked nuclei, representing follicular dendritic cells. (B) Blastoid variant. The nuclei have more open chromatin, resembling lymphoblastic lymphoma. Note the frequent mitoses. This tumor is TdT negative.")

The lymphoma cells often appear monotonous. They are slightly larger than small lymphocytes. Their nuclei show variable degrees of indentation and angulation, while usually maintaining an overall spheric configuration. Sometimes the nuclei can be perfectly round (see Figs. 21A.54A and 21A.55A ). The chromatin is fairly condensed, and nucleoli are inconspicuous. The mitotic count is variable, ranging from low to high. Cytoplasm is typically scanty. In some cases, some tumor cells may have an appreciable amount of pale cytoplasm, mimicking monocytoid B cells. Rare cases may show focal differentiation into mature plasma cells.
This variant is characterized by lymphoblast-like neoplastic cells. The cells have fine chromatin, and mitotic figures are readily identified (see Fig. 21A.55B ). A single biopsy may show areas with typical morphology and areas with blastoid appearance. Blastoid features may also appear in a subsequent biopsy of classic mantle cell lymphoma, and vice versa.
In the literature, this variant is sometimes included among the blastoid variant . The pleomorphic variant is characterized by heterogeneous populations of larger tumor cells with irregular nuclei and distinct nucleoli. There can be scattered large bizarre cells. This variant differs from large B-cell lymphoma in that the chromatin pattern is moderately condensed, nucleoli are often small, and cytoplasm is scanty; immunohistochemistry is essential for confirming the diagnosis ( Fig. 21A.57 ).

This is usually an incidental finding in reactive lymph nodes—cyclin D1+ small lymphoid cells are confined to the inner mantle zones of reactive follicles, without coalescence or spillover into interfollicular regions. The involved mantles may or may not be thickened. Distinction from reactive follicular hyperplasia is impossible without recourse to immunohistochemistry. In situ mantle cell neoplasia is very rare: In a study of 1292 reactive lymph nodes from unselected consecutive surgical specimens of 131 patients without a history of lymphoma, not a single case was found on immunostaining. In 29% of cases there is coexisting lymphoma of various types. Although data on long-term outcome are limited, in situ mantle cell neoplasia appears to have an indolent course, and long-term survival is the norm even without therapy. Progression to overt mantle cell lymphoma is rare; among 12 patients with a minimum follow-up of 1 year (median 3 years), 1 (8%) developed mantle cell lymphoma at 4 years. For this reason, the term in situ mantle cell neoplasia (as opposed to in situ mantle cell lymphoma ) is used in the 2017 WHO classification.
B-lineage markers (CD19, CD20, PAX5) are positive. The surface Ig is commonly of IgM ± IgD type, and the expression is stronger compared with CLL/SLL. Mantle cell lymphoma is unusual among B-cell lymphomas in that λ light chain is more commonly expressed than κ.
CD5+, CD23-, CD10-, BCL6-, cyclin D1+, SOX11+, LEF1-, CD200- ( Fig. 21A.58 ). Cyclin D1 expression is the hallmark of mantle cell lymphoma ( Fig. 21A.59A ); all other lymphoma types, with rare exceptions, are cyclin D1 negative. However, a small percentage of cases may deviate from this classic immunophenotype (e.g., up to 11% of cases can be CD5 negative, and up to 7% of cases can be cyclin D1 negative). For the cyclin D1 negative cases, upregulation of cyclin D2 or D3 (due to translocations involving their encoding genes) may substitute for cyclin D1, but immunostaining for cyclin D2 or cyclin D3 does not help in diagnosis because other types of lymphomas are also commonly positive for these markers. SOX11, a transcription factor not normally expressed in B cells, is a sensitive and specific marker for mantle cell lymphoma. Cyclin D1 negative mantle cell lymphomas are often positive for SOX11—a feature that facilitates the diagnosis. However, SOX11 staining is not entirely specific, but can also occur in Burkitt lymphoma (25%), lymphoblastic lymphoma (100%), and T-prolymphocytic leukemia (66%). Staining for LEF1 and CD200, in conjunction with SOX11, may aid in distinction from CLL/SLL in difficult cases; both markers are typically negative in mantle cell lymphoma and positive in CLL/SLL. In mantle cell lymphoma, there are commonly dispersed, and occasionally nodular, meshworks of FDCs interspersed throughout (see Fig. 21A.59B ). CD43 is commonly positive. There are often few admixed T cells.
 CD20 positive. (Middle) CD3 shows scattered reactive T lymphocytes among the tumor cells. (Right) The B cells (neoplastic cells) coexpress CD5, as evidenced by the presence of many more positive cells compared with the CD3 (T cell) stain.")
 The immunohistochemical hallmark is cyclin D1; note the nuclear staining. (B) Nuclear staining for SOX11. (C) Staining for CD21 typically highlights irregular loose meshworks of follicular dendritic cells.")
Exceptionally, CD8 expression has been reported.
Up to 7% of cases of mantle cell lymphoma may be negative for cyclin D1 by immunohistochemistry; the definitive diagnosis of such cases will rest on typical morphology, CD5 expression, and SOX11 expression. Cyclin D1 expression is also not pathognomonic of mantle cell lymphoma among hematolymphoid neoplasms either, because this can be seen in some cases of CLL/SLL (predominantly in proliferation centers), hairy cell leukemia, and myeloma/plasmacytoma. Rare cases of diffuse large B-cell lymphomas can be cyclin D1 positive, but they lack CCND1 gene translocation and lack immunoreactivity for SOX11.
IG genes are clonally rearranged. They are unmutated in 70% to 80% of cases and hypermutated in 20% to 30%. In contrast to CLL/SLL, there is no difference in clinical outcome between the unmutated and mutated groups. It has been suggested that mantle cell lymphoma may not be a pregerminal center (naïve) B-cell neoplasm as previously believed, but is derived from a marginal zone or peripheral blood memory B cell that has undergone extrafollicular T–independent antigen response.
Approximately 50% to 65% of cases exhibit the cytogenetic abnormality t(11; 14)(q13; q32), which juxtaposes the CCND1 oncogene on 11q13 (formerly known as BCL1 or PRAD1 ) with the IGH gene. The CCND1 gene encodes cyclin D1. Using the Southern blot technique, CCND1 gene rearrangement can be demonstrated in 50% to 60% of all cases. Using PCR, CCND1 gene rearrangement can be demonstrated in only 25% to 50% of cases. On the other hand, using FISH for 11q13 translocation or Northern blot analysis for cyclin D1 overexpression, practically all cases give positive results. As a result, FISH (using breakapart probes) is the preferred method for detecting CCND1 translocation in mantle cell lymphoma. In the rare cases of mantle cell lymphoma lacking CCND1 translocation, the most common genetic alteration is CCND2 gene fusion, often with IGK or IGL gene as partner.
Secondary cytogenetic alterations are very common: loss of chromosomes 13 and Y; deletions of 1p, 2p11, 6q, 8p, 11q22-23, 13q14, 13q34, and 17p; gains at 3q26-29 and 11q; and trisomy 12. Deletion involving chromosome 11q is found in 46% of cases, implicating ATM gene. Mutation of TP53 gene, deletion of P16 , loss of P16 expression, loss of P21 expression, and constitutive activation of the PI3K/Akt signaling pathway have been correlated with the blastoid/pleomorphic variant, which commonly shows tetraploidy, high-level DNA amplification, and high frequency of chromosomal imbalances. Up to 12% of cases harbor mutations in NOTCH1 , which is associated with poor overall survival.
Gene expression profiling studies show that mantle cell lymphoma exhibits a signature distinct from other low-grade B-cell lymphomas. The signature includes genes involved in apoptosis, cell cycle, signal transduction, and cell structures. There are also alterations in the tumor necrosis factor and NFκB pathways. Of interest, cyclin D1- cases are comparable to the cyclin D1+ cases with respect to expression of the signature genes; the survival rate is also no different. There is significant differential expression of some genes between the blastoid variant and classic mantle cell lymphoma, including tumor suppressors, transcription factors, protooncogenes, and genes associated with cell cycle regulation, proliferation, chromatin assembly, mitosis, and spindle assembly.
Clinical factors. Old age (>65–70 years) is associated with a worse prognosis. Peripheral blood involvement at diagnosis, splenomegaly, and high lactate dehydrogenase level are associated with a worse prognosis. Poor performance status, high IPI, high follicular lymphoma IPI (FLIPI), or high mantle cell lymphoma IPI (MIPI) is an unfavorable prognostic factor. The leukemic nonnodal variant is associated with an indolent clinical course.
Morphologic features. The blastoid/pleomorphic variant is associated with a worse outcome compared with classic mantle cell lymphoma. Nodular follicular dendritic cell pattern resembling that of primary follicles is associated with a more favorable prognosis.
Proliferation. A high mitotic count of above 20/10 high-power fields (hpf) or Ki67 index greater than 10% is associated with a worse outcome. High expression of proliferative signature genes can identify subgroups of patients that differ in median survival by more than 5 years.
Molecular features. V(H)321 usage is associated with a longer median survival, but the patients tend to be younger at diagnosis. Mutation of TP53 gene, overexpression of p53 protein, or deletion of P16 gene is correlated with a poor outcome. Based on the expression level of proliferation signature genes, patients can be further stratified into four prognostic groups with median survival times of 0.8, 2.3, 3.3, and 6.7 years, respectively. Use of quantitative reverse-transcription PCR to measure the expression of five genes ( RAN, MYC, TNFRSF10B, POLE2, SLC29A2 ), applicable to frozen or paraffin-embedded tissue, holds promise as a simpler method for survival prediction that can be performed in the diagnostic laboratory.
Other small B-cell lymphomas. Distinction from atypical CLL/SLL (CD23 negative) is often challenging and requires confirmation of cyclin D1 expression and/or CCND1 translocation. Exceptional cases of mantle cell lymphoma may show cytoarchitectural features mimicking extranodal marginal zone lymphoma of MALT.
Lymphoblastic lymphoma. Lymphoblastic lymphoma may mimic mantle cell lymphoma because of the irregular nuclear folding, but can be distinguished from the latter by the more delicate chromatin, higher mitotic count, high probability of T-cell immunophenotype, and younger age of the patients. The blastoid variant of mantle cell lymphoma is practically indistinguishable from lymphoblastic lymphoma but is TdT negative.
Mantle zone hyperplasia. Mantle zone hyperplasia sometimes affects a solitary node of young individuals. In contrast to mantle cell lymphoma with a mantle zone growth pattern, the follicles are localized to the cortex without architectural effacement, and there is no coalescence of the broadened mantles.
Castleman disease of hyaline vascular type.
Follicular lymphomas are tumors composed of follicular center B lymphocytes, which show a follicular organization.
Follicular lymphoma accounts for 22% to 40% of all NHLs in Caucasians but a much lower percentage (5%–10%) in Asian and developing countries. It occurs predominantly in middle-aged and old subjects (see Table 21A.9 ). The patients usually present with insidious painless enlargement of multiple lymph nodes, and there may be a history of waxing and waning of the nodes. Splenomegaly is common. Some patients have constitutional symptoms such as fever and malaise.
The disease is almost always in stage III/IV at presentation. Bone marrow involvement is common (30%–50% of cases) but does not influence the prognosis. In a proportion of patients, there are circulating lymphoma cells with notched or cleaved nuclei (buttock cells), so-called lymphosarcoma leukemia. Even in the absence of morphologic evidence of involvement, clonal populations can often be detected in the peripheral blood by flow cytometric or genotypic analysis. Furthermore, the clonal population often persists despite achievement of complete remission through treatment.
In the majority of patients, the disease pursues an indolent protracted course punctuated by multiple recurrences over many years. Despite a good response to radiotherapy and/or immunochemotherapy, recurrence is the rule; follicular lymphoma is practically incurable with currently available therapy, with the possible exception of stage I disease. The median survival is 5 to 10 years. Most patients will eventually succumb to the disease. Therefore treatment may be withheld in patients with no significant symptoms or life-threatening complications until there is evidence of tumor progression. Spontaneous regression can occur in up to 30% of patients, although the disease usually recurs eventually. Nonetheless, there is a small subset of patients whose disease progresses rapidly, often with transformation to aggressive lymphoma and early death.
Special types of follicular lymphoma (pediatric type, duodenal type, primary cutaneous, and in situ) are discussed separately in subsequent sections.
The nodal architecture is effaced by neoplastic follicles, some of which may extend into the perinodal tissue. Examination of histologic sections under very low magnification, ideally with reduced illumination, will facilitate appreciation of the follicular pattern. In most cases, the follicles are arranged in a back-to-back pattern with barely any interfollicular tissue ( Figs. 21A.60, 21A.61, and 21A.62 ). If further assessment confirms that the follicles are composed of follicular center cells, a diagnosis of follicular lymphoma can be made. In other cases, the follicles are discrete and well separated ( Fig. 21A.63 ). A diagnosis of follicular lymphoma is more difficult in these cases and requires consideration of other features (such as cytologic composition, presence or absence of mantles, tingible-body macrophages, and polarity) and immunohistochemical evaluation. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.



 presence of follicles outside the node (right upper field); (2) homogeneous appearance of the follicle centers (lack of tingible body macrophages and polarity); (3) predominance of centrocytes in the follicles when examined under higher magnification.")
The neoplastic follicles are usually round, but some can have very irregular contours. They vary in size but are usually smaller than reactive follicles. They are often poorly demarcated and vague, but mantle cuffs can be present or even thick ( Fig. 21A.64 ; see also Figs. 21A.61 and 21A.63 ). Neoplastic follicles lack the cellular polarization typically seen in reactive follicles. In most cases, tingible-body macrophages are absent or very sparse, but there can be exceptions (see Fig. 21A.64 ) .
 The follicles are closely packed. (Right) In this example, the follicles are less closely packed and thin mantles are present.")
The cellular composition of the follicles is variable. Cases composed predominantly of centrocytes (small cleaved cells) are the easiest to diagnose because reactive follicles do not show such a monotonous cellular composition. The centrocytes typically have folded, angulated, elongated, or triangular nuclei about twice the size of small lymphocytes. Their chromatin is dense, but less so than small lymphocytes. Cytoplasm is typically scanty ( Fig. 21A.65A ). Less commonly, the centrocytes exhibit a rounder contour and may simulate small lymphocytes. Occasional large blastic cells (centroblasts or large noncleaved cells) are always found. FDCs scattered within the neoplastic follicles can be distinguished from the blastic cells by their delicate violaceous nuclear membrane, empty chromatin, small nucleoli, and indistinct cellular boundaries.
 Grade 1, composed predominantly of centrocytes (small cleaved cells). Rare large cells are almost always found. Note absence of tingible-body macrophages. (B) Grade 2, composed of mixed small and large cells. An appreciable number of large blastic cells with distinct nucleoli and a definite amphophilic rim of cytoplasm are intermingled with the centrocytes (small cleaved cells). (C) Grade 3, composed predominantly of large cells.")
Follicular lymphomas composed of a mixed cellular population or predominantly large cells are less easy to diagnose because the cellular composition approximates that of reactive follicles (see Fig. 21A.65B ). The large cells (centroblasts or large noncleaved cells) have round or sometimes multilobated nuclei, vesicular chromatin, multiple membrane-bound nucleoli, and a definite rim of amphophilic to basophilic cytoplasm. Rare cells resembling Reed-Sternberg cells may be present.
In the interfollicular zone there are commonly atypical small to midsize lymphoid cells occurring singly, in clusters or sheets, indicative of involvement of this zone by lymphoma ( Fig. 21A.66 ). The nuclei of these atypical lymphoid cells are larger than those of small lymphocytes, and show irregular folding. Compared with the neoplastic cells within the follicles, they are often smaller and show a more rounded overall nuclear contour. This is one of the important features supportive of a diagnosis of follicular lymphoma versus reactive follicular hyperplasia.

Some degree of sclerosis is common, particularly in those cases occurring in the retroperitoneum and groin. The sclerosis is most prominent in the perinodal tissues and areas of diffuse growth. It can take the form of broad collagenous bands dividing the tumor into irregular nodules, or fine sclerosis resulting in compartmentalization of the lymphoma cells. Infiltration of the walls of the veins is another common and highly characteristic feature of follicular lymphoma ( Fig. 21A.67 ).
 A highly characteristic feature commonly observed in follicular lymphoma is invasion of the walls of veins by lymphoma cells. In contrast to the angioinvasion seen in extranodal NK/T-cell lymphoma, there is no fibrin deposit in or around the blood vessel wall, and there is usually no associated coagulative necrosis. (Right) Immunostaining for CD10 highlights the neoplastic population and provides support for the follicular center cell differentiation of the lymphoma cells.")
Bone marrow involvement characteristically occurs in a paratrabecular location, often accompanied by an increase in reticulin fibers. There may be variable involvement of the adjacent marrow. There are usually fewer large cells compared with the lymph node, suggesting that the smaller cells are more likely to enter the circulation.
The spleen is usually involved in a miliary fashion, with selective replacement of the white pulp by lymphoma. The red pulp, however, is also involved (see Chapter 21B ).
The most important element of grading of follicular lymphomas is the segregation of those that consist predominantly or entirely of large cells from those that do not, as the former group is more likely to show aggressive clinical behavior, with early relapse and high likelihood of progression to diffuse large B-cell lymphoma, but is more likely to respond to intensive chemotherapy. Many grading systems have been proposed, but the method devised by Mann and Berard has been most widely used and extensively tested, and is the basis for the WHO grading scheme. In this system, the number of large nucleolated cells is counted, using ×10 ocular and ×40 objective (0.159 mm 2 ). At least 10 fields of neoplastic follicles are counted, an average is taken, and three grades are assigned according to the count ( Table 21A.13 ). If the high power field size is not as indicated above, adjustments of the large cell count will be necessary. Of note, while formally retaining the three-tier system, the current WHO classification recommends use of a combined designation for grades 1 and 2, as the interobserver reproducibility for this distinction is poor and there are not meaningful differences in clinical behavior between these two groups, while the behavior and outcome for grade 3 follicular lymphoma (particularly grade 3b) is significantly different. This grading has been shown to correlate with survival, with a worse prognosis for those cases with more large cells, although the outcome may be altered significantly by the treatment scheme.
| Grade | Equivalent Terminology | Number of Large Nucleolated Cells Per High-Power Field, Based on Field Size of 0.159 mm 2 | |
|---|---|---|---|
| 1 a | Predominantly small cleaved cell (centroblastic-centrocytic) | ≤5 | |
| 2 a | Mixed small and large cells (centroblastic-centrocytic) | 6–15 | |
| 3 | 3a | Predominantly large cell (centroblastic-centrocytic) | >15, but there are still admixed centrocytes (small cleaved cells) |
| 3b | Large cell (centroblastic) | Exclusively large nucleolated cells | |
| Growth Pattern | % Area With Follicle Formation |
|---|---|
| Follicular | >75% |
| Follicular and diffuse | 25%–75% |
| Focally follicular/predominantly diffuse | <25% |
| Diffuse | 0% |
Grade 3b follicular lymphoma is a special category in which the neoplasm is composed exclusively of centroblasts (large noncleaved cells), without admixed centrocytes. In contrast with grade 3a follicular lymphoma, CD10 expression is less frequent (50% vs 100%), t(14; 18) is uncommon (22% vs 73%), and other chromosomal abnormalities are common, such as 3q27 translocation, +1/1q, del(6q), and +7/7p. That is, it appears to be biologically more related to DLBCL. Although there is no difference in survival between grade 3b and grade 3a follicular lymphoma, the distinction is critical due to its impact on treatment: Upfront multiagent chemotherapy, including rituximab, is the norm for grade 3b follicular lymphoma, while a watch-and-wait strategy, similar to that for low-grade disease, may still be appropriate in selected asymptomatic patients with newly diagnosed grade 3a follicular lymphoma.
Grading of follicular lymphoma dominated by large cleaved cells/centrocytes (diameter comparable to centroblasts) is currently unclear. These cases are commonly CD10+ and BCL2+, with high Ki67 index and BCL2 rearrangement.
In follicular lymphoma, there can be a diffuse component, defined by an area of lymphomatous infiltrate completely devoid of neoplastic follicles. If there is uncertainty, immunostaining for CD21 and/or CD35 is helpful to confirm genuine absence of FDC meshworks in the area ( Fig. 21A.68 ). A broad interfollicular zone does not qualify for a diffuse component. (The terminology used to quantify the diffuse component is detailed in Table 12A.13. )
, and absence of follicular dendritic cell meshworks in the diffuse component (left field).")
Presence of diffuse areas is generally considered a progression phenomenon in follicular lymphoma, although its impact on clinical outcome is controversial. For grade 1 and 2 follicular lymphomas, the prognosis is worsened only if the diffuse areas constitute at least 25% to 50% of the tumor. However, for grade 3 follicular lymphoma, any diffuse component may worsen the prognosis; in fact, such cases should be designated DLBCL with follicular lymphoma instead of follicular lymphoma, grade 3, follicular and diffuse .
Approximately 9% of follicular lymphomas are accompanied by a prominent neoplastic marginal zone component comprising cells similar to marginal zone or monocytoid B cells ( Figs. 21A.69 and 21A.70 ). In contrast to the neoplastic follicle centers, the marginal zone cells do not express CD10 and BCL6. While some studies suggest the presence of more than 5% marginal zone B cells in follicular lymphoma to be associated with advanced stage disease (87%), bone marrow involvement (69%), and a poor 5–year failure-free survival (17%), other studies have not shown this feature to be of prognostic significance.
 The neoplastic follicle centers are surrounded by broad pale-staining haloes formed by cells with features of monocytoid B cells. (Right) CD10 immunostaining selectively highlights the neoplastic follicle centers, but not the cells in the marginal zone.")

Some follicular lymphomas are composed partly or entirely of signet ring cells, with the nuclei displaced to one side by a clear vacuole or eosinophilic PAS-positive globule (Russell body type) ( Fig. 21A.71 ). The clear vacuoles often stain for Ig (usually IgG) in the rim and are shown ultrastructurally to be composed of even-sized spherules or irregular electron-dense clumps. The eosinophilic globules usually stain for IgM and are shown ultrastructurally to be amorphous electron-dense material within distended rough endoplasmic reticulum.
 The nucleus is eccentrically displaced by an empty cytoplasmic vacuole. This variant is often of IgG type. (B) In this variant (usually of IgM type) the nucleus is displaced by an eosinophilic cytoplasmic globule.")
Some follicular lymphomas contain abundant extracellular PAS-positive amorphous eosinophilic precipitate, shown ultrastructurally to be composed of membrane-bound vesicles and electron-dense bodies. Since these are membranous structures, they stain for various B–lineage surface markers and Ig.
The rosettes are formed by neoplastic lymphocytes arranged around eosinophilic fibrillary material. Since the fibrillary material represents entangled cytoplasmic processes, it stains with the various B–lineage markers.
When the mantle zone small lymphocytes show prominent inward extension into the neoplastic germinal centers resulting in flowerlike appearance, the pattern can mimic progressive transformation of germinal centers ( Fig. 21A.72 ). Absence of reactive follicles in the background, the cytologic composition, and involvement of perinodal tissues favor a diagnosis of follicular lymphoma.
 The follicles are large and dark staining. (Right) Tongues of lymphocytes extend into the follicles, producing a floral appearance.")
Rare follicular lymphomas show reversal of the normal zonation, with the centers of the follicles being composed mostly of dark-staining small cells and the rims composed of paler larger cells ( Fig. 21A.73 ). This variant differs from follicular lymphoma with marginal zone differentiation in that the cells in both zones are CD10+ and BCL6+.

The neoplastic cells have highly irregularly folded cerebriform nuclei, resembling those of mycosis fungoides.
Immunoblasts and plasmablasts may constitute the major cellular composition of some follicular lymphomas.
In some follicular lymphomas, a plasma cell population staining for the same Ig light chain as the neoplastic follicles is present in the interfollicular areas or within the follicles ( Fig. 21A.74 ). Some such cases are associated with paraproteinemia. In rare cases, the neoplastic follicles are composed exclusively of plasma cells (follicular plasmacytoma).

The neoplastic follicles may rarely be associated with penetrating hyalinized vessels, mimicking hyaline-vascular Castleman disease.
In this variant, the follicles are stuffed with numerous small T cells, which overshadow the minor component of neoplastic follicular center B cells. On closer scrutiny, the CD3+ cells are small lymphocytes, while the CD20+ cells are larger, and there are also areas where CD10+ cells spill over into the interfollicular zone.
In situ follicular neoplasia (formerly follicular lymphoma in situ ) is characterized by partial or total colonization of reactive germinal centers by BCL2 -rearranged clonal B cells. It can occur in lymph node or at extranodal sites. Lymph node shows usual reactive follicular hyperplasia, with some germinal centers exhibiting variable replacement by tight clusters of centrocytes. Immunostaining reveals aggregates of intensely BCL2+ and intensely CD10+ cells within some germinal centers ( Fig. 21A.75 ). There is no evidence of interfollicular infiltration. Molecular studies can confirm that the BCL2+ follicles are clonal and exhibit BCL2 gene rearrangement, while the admixed BCL2– follicles are polyclonal.
. Most follicles are BCL2 negative or contain small numbers of intensely BCL2 positive cells.")
The patients are generally otherwise asymptomatic. Since some patients may have synchronous follicular lymphoma or other types of lymphoma (including Hodgkin and non-Hodgkin types) at another site, proper staging is essential. If staging is negative, the risk of progression to overt lymphoma is very low (~5%). Therefore treatment is currently not recommended.
This is presumably a preneoplastic process in which the full complement of genetic alterations necessary for malignant transformation has not been acquired. This can be considered the tissue equivalent of the circulating, nontransformed B cells harboring a t(14; 18) that can be detected in normal populations with sensitive methods. The incidence of in situ follicular neoplasia in otherwise reactive-appearing lymph nodes is very low, approximately 2%. Thus it is neither necessary nor cost effective to perform BCL2 immunostaining on a routine basis for all reactive-looking lymph nodes.
An important differential diagnosis is partial lymph node involvement by follicular lymphoma, characterized by the presence of crowded and enlarged follicles with attenuated or disrupted mantles in a portion of a lymph node. There is often evidence of infiltration of the tissues outside the follicles. That is, the morphology is just like conventional nodal follicular lymphoma, except that the nodal involvement is focal. BCL2 or CD10 immunostaining of the germinal centers is variable and is not necessarily intensely positive. Some patients are found on staging to have follicular lymphoma elsewhere. For those with negative staging, follow-up will reveal overt follicular lymphoma in a high proportion of patients (53%).
B-lineage markers are positive. The usual Ig isotype is IgM, but a second isotype can be expressed (IgG, IgD, or IgA). Some tumors are Ig negative, particularly those composed mostly of large cells.
BCL2+, CD10+, BCL6+, HGAL+, LMO2+, STMN1+, CD5-, cyclin D1-. The normal follicular center B cells are BCL2 negative, while BCL2 is expressed in the neoplastic follicles in approximately 85% of cases ( Fig. 21A.76 ). The positivity rate is lower for cases composed predominantly of large cells. Negative staining for BCL2 may result from lack of t(14; 18) or presence of mutations in the translocated BCL2 gene. To address the latter problem, it is recommended that BCL2 negative cases be stained with an alternative BCL2 antibody (such as E17) to prevent false negativity. Follicular center cell markers include CD10, BCL6, HGAL, STMN1 (stathmin), and LMO2, but it is not necessary to perform immunostaining for all of them in the routine workup of suspected follicular lymphoma. FDCs, which form tight or distorted meshworks, can be demonstrated within the neoplastic follicles. The interfollicular tissue is rich in T cells, but when dominated by B cells is indicative of involvement of the interfollicular zone by follicular lymphoma. The Ki67 (proliferation) index is variable but often much lower than that of reactive follicular hyperplasia (mean 15.6% vs 64.9%) (see Fig. 21A.76 ).
 Reactive follicular hyperplasia: Only the mantle zone and interfollicular cells are BCL2 positive, while the follicle centers are negative except for scattered cells, which represent intrafollicular T cells. (B) Reactive follicular hyperplasia: Ki67 immunostaining shows up the polarity of the follicles as well as the extremely high proliferative activity . (C) Follicular lymphoma: The follicle centers are positive for BCL2. (D) Follicular lymphoma: Ki67 proliferative fraction is often lower than that of reactive follicular hyperplasia. There is commonly concentration of positive cells in the peripheral portion of the follicle centers.")
Rare cases of follicular lymphoma express CD5, in particular the floral variant. CD43 is often negative. CD23 is expressed in a proportion of cases. T lymphocytes, usually of CD4+ subset, are scattered within the neoplastic follicles. Reactive CD57+ cells are frequently scattered in the neoplastic follicles and sometimes in the interfollicular regions.
The clonally rearranged IG genes show somatic hypermutation and ongoing mutations.
Follicular lymphoma characteristically shows t(14; 18)(q32; q21), whereby the BCL2 gene on chromosome 18 is juxtaposed to the IGH gene on chromosome 14. This translocation is seen in 70% to 90% of cases. The translocation results in overexpression of a structurally normal BCL2 protein in the neoplastic follicle center cells. Since BCL2 protein plays an important role in blocking apoptosis, the neoplastic cells have a survival advantage. FISH is the most sensitive technique for detection of t(14; 18), while PCR may give false negative results. t(14; 18) is also present in some DLBCLs; such cases are believed to have transformed from an occult follicular lymphoma. In the small proportion of cases of follicular lymphoma that lack t(14; 18) but show BCL2 protein expression, gains in 18q (increased copies of BCL2 gene) are commonly found.
For follicular lymphomas lacking t(14; 18), the gene expression and microRNA profiles are distinct from those with t(14; 18). These cases show a late germinal center B-cell phenotype. At least a proportion of t(14; 18) negative follicular lymphomas show rearrangements of the BCL6 gene, a feature found in 5% to 15% of follicular lymphomas. This is correlated with grade 3b histology, high proliferative index, and infrequent expression of CD10 and BCL2. Some follicular lymphomas can show rearrangements of both BCL2 and BCL6 . Somatic mutations in the BCL6 gene occur in 47% of cases and result from the general phenomenon of non– IG gene hypermutation in cells of the follicular center.
In follicular lymphoma, recurrent mutations have been found in the histone methyltransferases MLL2 (89%) and EZH2 (7%), and the histone acetylases CREBBP (33%), EP300 (9%), and MEF2B (15%).
A small percentage of cases (~2%) may be positive for EBV. The positive cases occur in patients with no evidence of immunodeficiency and often show grade 3 histology.
Patient-related factors. Old age (>70 years), poor performance status, and male gender are associated with a worse prognosis.
Prognostic indices. Some studies have shown that FLIPI can better stratify patients with follicular lymphoma into different prognostic groups compared with IPI. Five adverse prognostic factors are included in the index: age (>60 years vs ≤60 years), Ann Arbor stage (III-IV vs I-II), hemoglobin level (<12 g/dL vs ≥12 g/dL), number of nodal areas (>4 vs ≤4), and serum lactate dehydrogenase (LDH) level (above normal vs normal or below). Three risk groups are defined: low risk (no or one adverse factor), intermediate risk (two factors), and poor risk (three or more adverse factors). The Italian Lymphoma Intergroup (ILI) prognostic index, which takes into account age, gender, B symptoms, number of extranodal sites, erythrocyte sedimation rate (ESR), and LDH level, has also been reported to be more discriminatory than IPI in identifying patients with different survivals.
Tumor extent. Patients with limited stage disease have a better prognosis. High tumor burden, reflected by lymph node size, number of extranodal sites, degree of marrow involvement, degree of hepatosplenomegaly, and presence of circulating lymphoma cells is of prognostic relevance. Low hemoglobin level, elevated ESR, elevated LDH level, and high level of β2–microglobulin, which are surrogate laboratory markers for high tumor load, are correlated with lower response rate to treatment and shorter survival.
Histologic grade and proliferative index. Patients with grade 1 or 2 follicular lymphoma have significantly longer overall survival than those with grade 3 follicular lymphoma, while the disease-specific survival is not statistically different. Among grade 1 or 2 follicular lymphomas, there is a subgroup showing a discordantly high proliferative index (Ki67 index ≥30%)—the overall survival curve shows a rapid drop-off in the first few years and then attains a plateau, and the disease-specific survival is significantly worse than that of usual grade 1 or 2 follicular lymphoma. Thus the clinical behavior of this subgroup appears to be more akin to grade 3 follicular lymphoma.
Microenvironment. A high content of lymphoma-associated macrophages is an independent predictor of poor survival. A high number of FOXP3+ regulatory T cells is correlated with better survival.
Additional genetic changes. Presence of genomic aberrations other than t(14; 18), in particular 6q and 17p deletions, is correlated with shorter survival and higher probability of transformation to DLBCL.
Gene expression profiling. According to one study, a gene expression profile of 81 genes can distinguish low-grade from high-grade follicular lymphoma. This stratification profile is reported to be a more reliable predictor of clinical behavior than the currently used histologic grading and clinical criteria. According to another study, the length of survival among patients with follicular lymphoma correlates with the molecular features of tumor-infiltrating immune cells, independent of clinical prognostic variables.
Reactive follicular hyperplasia. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
CLL/SLL. See “Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma.”
Lymphoblastic lymphoma. See “Lymphoblastic Lymphoma.”
NLPHL. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Follicular lymphoma is a dynamic and unstable neoplasm, which can take up different appearances in different sites and at different times. Biopsy of one lymph node may show predominance of small cells and pure follicular growth, while another biopsy may show a mixed cell composition with a follicular and diffuse pattern. Some patients initially diagnosed as having DLBCL may be found, after treatment, to have follicular lymphoma. It is likely that these patients had ab initio follicular lymphoma, which has transformed into a large cell lymphoma at the time of presentation. The former component becomes evident as the latter more chemosensitive component is eradicated by treatment. Alternatively, some patients with DLBCL are found on staging (such as marrow examination) to have an underlying follicular lymphoma (i.e., so-called discordant involvement).
Since BCL2 expression inhibits apoptosis, the lymphoma cells are long lived, permitting additional genetic alterations to accumulate. Such changes can lead to progression of the follicular lymphoma, taking the form of diffuse growth, transformation to large cell lymphoma, and blastic transformation.
Follicular lymphoma can evolve into a diffuse growth phase without a change in cytologic composition. This phenomenon does not necessarily worsen the prognosis, especially if the diffuse component is minor. However, if the follicular lymphoma is of large cell type (grade 3), essentially the diffuse component has to be diagnosed as DLBCL, and the prognosis is worsened.
Transformation to DLBCL may occur, at a frequency of more than 50% on long follow-up. The transformation appears to be part of the natural history of follicular lymphoma, rather than a consequence of the mutagenic effects of chemotherapy or radiotherapy. It occurs at 7 to 300 months after diagnosis of follicular lymphoma. In general, transformation is associated with an aggressive clinical course, but this tumor is paradoxically amenable to cure with long-term disease-free survival in a proportion of patients treated aggressively, a level of success similar to de novo DLBCL treated with aggressive chemotherapy. Modern regimens of multiagent chemotherapy have significantly improved outcomes in transformed follicular lymphoma, with reported median overall survival of 5 years posttransformation; favorable prognostic factors include limited disease, no prior chemotherapy, and complete response to therapy. The large cell lymphoma usually has the appearances of conventional DLBCL but can have an anaplastic appearance with sinusoidal distribution and CD30 expression. A number of genetic changes can mediate the transformation, such as TP53 gene mutation, somatic mutation in the translocated BCL2 gene, RAS mutation (rare), MYC rearrangement, and P16 deletion or inactivation. Of interest, the gene expression profile of transformed DLBCL is more similar to the antecedent follicular lymphoma than to de novo DLBCL.
Rare examples of transformation to lymphoma resembling Burkitt lymphoma have been reported. Many of these cases show MYC translocation in addition to the preexisting BCL2 rearrangement (so-called double-hit lymphoma), and the recommended terminology for the transformed lymphoma is high-grade B-cell lymphoma with MYC and BCL2 rearrangements . Lymphoblastic transformation (i.e., to a lymphoma resembling lymphoblastic lymphoma) may also occur; these are also typically associated with MYC rearrangement, while TdT immunoreactivity is variable. The clinical course is often rapidly progressive.
There exists a diffuse form of follicular center cell lymphoma composed of centrocytes (small cleaved cells) admixed with some centroblasts (large noncleaved cells—i.e., grade 1 or 2) ( Fig. 21A.77 ). This entity is uncommon, and the diagnosis requires confirmation by immunohistochemistry (expression of germinal center cell markers such as CD10, BCL6, and HGAL, and absence of follicular dendritic cell meshworks on CD21 staining) and/or genetic studies ( BCL2 gene rearrangement). Expression of CD23 is common. In all cases, in particular when the diagnostic material is limited (such as core biopsy), the possibility of biopsy from the diffuse component of an otherwise typical follicular lymphoma should also be considered.
.")
Although 1p36 aberrations are common in classical t(14; 18)+ follicular lymphoma, there exists a distinctive type of nodal follicular lymphoma lacking t(14; 18) and showing 1p36 abnormalities, although this type is currently not recognized in the revised 4th edition of WHO classification. The patients often present with low clinical stage disease, manifesting as large inguinal lymph nodes. The follicular lymphoma is diffuse or predominantly diffuse, usually grade 1 or 2. Germinal center cell markers and CD23 are expressed, while BCL2 is negative. Characteristic genetic changes are lack of BCL2 rearrangement and the presence of 1p36 deletion or TNFRSF14 (located on 1p36) mutations. The mutational landscape is distinct from classic follicular lymphoma, with frequent mutations in STAT6, CREBBP, and basal membrane protein genes.
A rare form of follicular lymphoma affecting pediatric patients and young adults (typically between the ages of 5 and 25) is recognized as a distinct entity. The distinctive features include the following :
There is a marked male predominance (M:F ratio >10 : 1).
Most patients have stage I/II disease.
Involvement of the head and neck region is commonest (such as cervical lymph node).
Most cases are of mixed or large cell type (grade 2 or 3) and show a high proliferative index (Ki67 >30%).
BCL2 immunostaining is usually negative, and BCL2 gene rearrangement is absent.
CD10 is strongly positive, often confined to the follicle centers (without spillover to interfollicular zone).
Prognosis is excellent. The disease is apparently curable, with recurrence or progression being very rare.
Morphologically, the follicles are large and expansile, comprising a high content of large cells that often show blastoid morphology (grade 3) and high mitotic count. Mantle zones are thin or absent ( Fig. 21A.78 ). Because of the morphologic resemblance to florid follicular hyperplasia, it is prudent to demonstrate the presence of clonal IG rearrangements before rendering a diagnosis of pediatric-type follicular lymphoma. The tumor shows low genetic complexity, with the most common changes being copy number neutral loss of heterozygosity of 1p36 locus, TNFRSF14 mutations/deletions (29%–54% of cases), MAP2K1 activating mutations (43%–49%), and KMT2D mutations (16% of cases).
 The lymph node shows irregular-shaped expansile follicles with incomplete thin mantles. Residual nodal tissue is evident in the left upper field. (B) The follicles comprise predominantly blastoid midsize lymphoid cells, accompanied by tinigble-body macrophages.")
Although pediatric-type follicular lymphoma most frequently affects children and young adults, it may also be seen rarely in older adults. Conversely, in adolescents and young adults, conventional adult-type follicular lymphomas can occur, and they can be recognized by BCL2 immunoreactivity, presence of t(14; 18), and a low mitotic index. Such cases are associated with high stage disease at presentation with clinical outcome similar to conventional adult-type follicular lymphoma.
Duodenal-type follicular lymphoma is a distinctive variant of follicular lymphoma. It most commonly presents as isolated or multiple tiny polyps in the second part of the duodenum, although similar lesions are also commonly simultaneously present in the jejunum and ileum. Patients are usually asymptomatic middle-aged adults, whose polyps are discovered incidentally during endoscopic examination for various reasons. The lymphoma involves the mucosa and/or submucosa, consisting of crowded or noncrowded follicles comprising predominantly centrocytes (grade 1, sometimes grade 2), lacking tingible-body macrophages and polarity ( Fig. 21A.79 ). Similar atypical cells are also found outside the follicles, and thus this is not a form of in situ follicular neoplasia. The immunophenotype and genotype are similar to those of conventional follicular lymphoma (i.e., positive for B-lineage markers, germinal center cell markers, BCL2, and t[14; 18] with BCL2 gene rearrangement). In contrast to the latter, the disease is localized rather than systemic, and the outcome is excellent with limited chemotherapy, rituximab therapy, localized radiation, or observation alone. Dissemination outside the intestines is very rare.
 are present in the duodenal mucosa. The lamina propria is also involved, being packed with dark-staining lymphoid cells.")
Follicular lymphoma sometimes occurs as localized disease in the skin, most commonly scalp, forehead, and trunk. The prognosis is excellent, with 5-year survival over 95%. Dissemination is uncommon, occurring in only about 10% of cases.
Histologically the pattern of skin involvement can be follicular, follicular and diffuse, or diffuse. The infiltrate consists of small, midsize, or large centrocytes admixed with variable numbers of centroblasts. These cells are positive for B-lineage markers, BCL6, and variably CD10. In contrast to conventional follicular lymphoma, BCL2 immunoreactivity and BCL2 rearrangement occur at a low frequency. The differential diagnosis of DLBCL of leg type may be raised for cases showing a diffuse growth pattern and predominance of large cells. The major distinguishing features are (1) large cells are mostly large centrocytes (without conspicuous nucleoli) rather than centroblasts (round nuclei with conspicuous nucleoli), and (2) BCL2-, MUM1- immunophenotype.
Testicular follicular lymphoma is considered a distinctive variant of follicular lymphoma in the revised 4th edition of WHO classification. It most frequently occurs in children, although it may rarely be seen in adults. In contrast to nodal follicular lymphoma, testicular follicular lymphoma is frequently of high histologic grade (usually grade 3A) and lacks evidence of BCL2 translocation. Prognosis is excellent, and surgical excision alone may be sufficient therapy.
A subset of follicular lymphomas occurring in the thyroid, spleen, and ovary exhibits features very similar to testicular follicular lymphoma. Follicular lymphomas occurring in these sites fall into two groups: (1) classic follicular lymphoma with BCL2 rearrangement, usually low histologic grade, and high clinical stage; or (2) follicular lymphoma without BCL2 rearrangement, usually grade 2 or 3, low clinical stage, and highly favorable prognosis.
Extranodal marginal zone lymphoma of MALT, also known as MALT lymphoma, is an indolent lymphoma showing architectural and cytologic homology with the normal lymphoid tissues occurring in various mucosal and extranodal sites.
Extranodal marginal zone lymphomas can occur in a wide variety of extranodal sites, in particular the gastrointestinal tract, thyroid, salivary gland, and lung, and less commonly, the ocular adnexa, thymus, skin, soft tissue, breast, tongue, tonsil, gall bladder, liver, urogenital tract, and dura. They can affect any age group of either sex (see Table 21A.9 ). The patients present with a mass lesion or nonspecific symptoms; some are incidentally found to harbor the lymphoma. Known predisposing conditions are chronic infection/inflammation (such as Helicobacter -associated gastritis) and autoimmune diseases (such as Hashimoto thyroiditis and Sjogren syndrome).
Since the lymphoma tends to be localized to the involved organ and/or regional lymph nodes for a long time before dissemination occurs, a high proportion of cases can be treated satisfactorily by locoregional therapy. However, there can be synchronous or metachronous involvement of more than one mucosal site, a phenomenon attributed to the homing properties of mucosal lymphocytes. Studies suggest that local or systemic dissemination occurs more frequently than was originally believed. On thorough staging, 25% of patients with gastric marginal zone lymphoma have multiorgan involvement, with dissemination beyond the gastrointestinal tract in 40% of these patients. Significantly more patients with extragastric marginal zone lymphoma show dissemination to another mucosa-associated lymphoid tissue organ (46%). Bone marrow involvement is reported in 2% to 15% of cases. Multifocality is significantly correlated with t(11; 18)(q21; q21) in gastric lymphomas and with trisomy 18 in extragastric lymphomas. Nonetheless, there is no significant difference in survival between patients with localized and disseminated disease.
The prognosis is highly favorable, although late local or distant relapses can occur. The 5-year overall survival according to the Non-Hodgkin Lymphoma Classification Project is 74%, and the figure for the gastric cases is more than 90%. The relapse rate is 37% (22% for gastric tumors and 48% for extragastric tumors), and the median time to relapse is 47 months.
In 9% to 30% of cases of extranodal marginal zone lymphoma, a large cell lymphoma can supervene, which will worsen the prognosis. Rare cases of composite extranodal marginal zone lymphoma and classic Hodgkin lymphoma have been reported.
The most dramatic paradigm change in the treatment approach to gastric extranodal marginal zone lymphoma has been the use of antibiotics. Approximately 70% to 80% of gastric extranodal marginal zone lymphomas associated with Helicobacter gastritis show complete remission with anti– Helicobacter therapy. Regression of the lymphoma usually occurs within a few weeks, but may take up to 1 year. Antibiotic therapy appears to be effective predominantly in pure low-grade disease confined to the mucosa and submucosa, although recent studies suggest that even gastric large B-cell lymphomas (early stage) show complete remission with anti- Helicobacter therapy in a high proportion of cases. Eradication of Helicobacter suppresses the Helicobacter -associated mucosal T-cell proliferation, which is required to provide contact-dependent help to the neoplastic B cells via CD40 and CD40–ligand interaction, leading to regression of the neoplasm. PCR-based studies show that molecular response lags behind morphologic response by 1 to 2 years, and molecular evidence of tumor may persist in some patients. Despite the clinical and histologic evidence of remission, it is likely that some residual tumor cells remain dormant, and they have the potential to regrow quickly upon restimulation as evidenced by a few reports of rapid recurrence on reinfection by Helicobacter . Of interest, patients with early stage gastric marginal zone lymphoma negative for Helicobacter may still benefit from antibiotic treatment as the sole treatment modality. On the other hand, Helicobacter eradication therapy is ineffective for extragastric extranodal marginal zone lymphomas.
Extranodal marginal zone lymphoma shows unifocal or multifocal involvement of the extranodal tissue. Reactive lymphoid follicles are frequently interspersed throughout. The neoplastic lymphoid infiltrate is diffuse, interfollicular, or perifollicular ( Fig. 21A.80 ). The infiltrate often comprises a mixture of cell types, although rare cases can exhibit a monomorphic population ( Fig. 21A.81 ). The most characteristic cell type is centrocyte-like, being small to midsize, with slightly folded or elongated angulated nuclei, moderately dense chromatin, inconspicuous nucleoli, and a small amount of pale to clear cytoplasm (see Fig. 21A.81 ). Some cells are slightly larger, with abundant clear cytoplasm, resembling monocytoid B cells (see Fig. 21A.81A ). Some cells possess dark round nuclei, indistinguishable from small lymphocytes. There are often admixed large cells resembling centroblasts or immunoblasts.

 Note the typical lymphoepithelial lesions formed by expansion and destruction of the gastric glands by lymphoma cells. The lymphoma cells are centrocyte-like, with clear cytoplasm. Note the mixture of cell types, including plasma cells. (B) In this case, there is a monotonous population of small cells.")
There is a tendency for the lymphoma cells to invade the epithelial structures, forming lymphoepithelial lesions (see Fig. 21A.81A ), which are defined as epithelial units expanded or distorted by groups of more than three lymphoid cells. With time, the destroyed epithelium may remain as isolated or small groups of oncocytic or signet ring cells ( Fig. 21A.82A ). Of interest, the lymphoma cells within and around the epithelial structures often assume a monocytoid B–cell–like appearance, producing pale collars around the lymphoepithelial lesions, a feature most strikingly observed in the salivary gland ( Fig. 21A.83 ). While lymphoepithelial lesions are characteristic of extranodal marginal zone lymphoma, they are not pathognomonic, and can be seen sometimes in other lymphoma types or reactive lymphoid proliferations.
 There is marked destruction of the glands, and residual epithelial cells have an oncocytic appearance or ground-glass appearance. In a biopsy specimen, this feature can potentially lead to a misdiagnosis of diffuse-type gastric carcinoma. (B) This biopsy shows the typical changes of lymphoma regression after anti- Helicobacter therapy. There is loss of glands due to prior destruction by lymphoma. The lamina propria is formed by loose fibrovascular tissue with few lymphoid cells—these spaces were once occupied by lymphoma cells, which have now disappeared.")

Plasma cells are often intermingled with the neoplastic infiltrate or are found beneath the surface epithelium. They are monotypic in about one-third of cases and therefore represent part of the neoplastic clone. In some cases, plasma cells are so prominent that distinction from an extraosseous plasmacytoma is difficult. The plasma cells can exhibit atypical features such as enlarged nuclei, distinct nucleoli, cytoplasmic crystals, and Dutcher bodies. Sometimes there can be extracellular Ig or amyloid deposits.
Follicular colonization is a common phenomenon. The neoplastic cells invade and replace the reactive follicles, resulting in vague or discrete nodular structures mimicking follicular lymphoma. The neoplastic cells that have colonized the follicles may show increased plasma cell differentiation or a more blastic appearance.
The regional lymph nodes are involved in approximately 30% of cases. In the early phase, the involvement is confined to the marginal zones around reactive follicles. With time, there is coalescence of the broadened marginal zones, colonization of the follicles, and interfollicular invasion resulting in architectural effacement. On the rare occasions of splenic involvement, the marginal zone is preferentially involved, with intact reactive germinal centers and mantle zones in the white pulp follicles.
After completion of anti- Helicobacter therapy, gastric biopsies are usually taken at 6 weeks to (1) confirm eradication of Helicobacter, and (2) assess the histologic response of the lymphoma. Thereafter the status of the lymphoma will be monitored at intervals of 3 to 6 months. Currently the gold standard for assessment of response to anti- Helicobacter therapy is still histologic rather than molecular study. The characteristic change of regressed lymphoma is a hypocellular lamina propria comprising loose connective tissue with scattered plasma cells, devoid of organized collections of lymphoid cells (see Fig. 21A.82B ). The empty-looking stroma results from disappearance of lymphoma cells and prior destruction of the glands by the lymphoma.
B-lineage markers are positive. Ig is usually of IgM, sometimes IgG or IgA type, but not IgD.
CD5-, CD10-, CD23-, cyclin D1-, BCL6-. Since there are no positive defining markers to aid in the diagnosis of marginal zone lymphoma, this category risks becoming a wastebasket for unclassifiable cases of low-grade B-cell lymphoma with no distinctive morphologic and immunophenotypic features. Immunoglobulin superfamily receptor translocation-associated 1 (IRTA1), which recognizes the marginal zones in human lymphoid tissues other than the spleen, is reported to be a promising marker for marginal zone lymphomas. IRTA1 expression is found in 93% of extranodal and 73% of nodal marginal zone lymphomas, as well as in lymphomas with marginal zone differentiation, but not in other types of lymphomas. There is aberrant nuclear expression of BCL10 for cases that show t(11; 18) or t(1; 14) translocation; in the stomach, this feature is correlated with a lack of response to anti- Helicobacter therapy.
BCL2 is often positive, and CD43 expression is observed in approximately 50% of cases. The Ki67 (proliferation) index is low.
Rare cases can be CD5+. While it has been suggested that CD5 expression in extranodal marginal zone lymphoma has a propensity for marrow involvement, this is not always the case.
The IG genes are clonally rearranged, with somatic hypermutations. There is also evidence of ongoing mutation in the IG genes, suggesting that antigen stimulation plays a role in clonal expansion. Rearrangements of the CCND1 , BCL2, and BCL6 genes are typically absent. Several distinctive but mutually exclusive chromosomal translocations are recognized in this lymphoma type, and not found in nodal or splenic marginal zone lymphomas. They occur at highly variable frequencies in different extranodal sites. Of interest, these disparate translocations all result in constitutive activation of NFκB (which transactivates genes that are important for cellular activation, proliferation, and survival) :
t(11; 18)(q21; q21), which results in fusion of BIRC3 (formerly known as API2, and an inhibitor of apoptosis) with MALT1 , occurs in 18% to 35% of cases. The resulting chimeric protein can directly activate NFκB, bypassing the normal signaling sequence. Absence of t(11; 18) in extranodal marginal zone lymphoma with a large cell component or large B-cell lymphoma suggests that the translocation may confer immunity against transformation. This translocation is found predominantly in tumors of the stomach and lung, and is uncommon in other extranodal sites. Tumors carrying t(11; 18) tend to show a relatively monotonous lymphomatous infiltrate; in the stomach, this translocation is correlated with a lack of response to anti-Helicobacter therapy.
t(1; 14)(p22; q32), which occurs in 1% to 2% of cases, is associated with more disseminated or aggressive tumor. The translocation juxtaposes the BCL10 to IGH gene, causing overexpression of wild-type BCL10 protein. BCL10 protein is believed to form oligomers without the need of upstream signaling, binds MALT1, and mediates its oligomerization, and subsequently triggers the molecules that lead to NFκB activation.
t(14; 18)(q32; q21), which occurs in about 15% of cases, juxtaposes MALT1 with IGH gene, resulting in overexpression of MALT1 protein. This translocation is found predominantly in tumors of the salivary gland, eye, skin, and lung. Additional genetic aberrations are common, such as trisomy 3 and/or 18.
t(3; 14)(p14.1; q32), which occurs in about 10% of cases, results in fusion of IGH and FOXP1 . This translocation is found predominantly in tumors of the thyroid, eye, and skin.
Other common genetic changes are trisomy 3, 12, and 18; loss of heterozygosity or mutation of TP53; P15 and P16 promoter methylation; and FAS gene mutation. Trisomy 3 is found in approximately 60% of cases by chromosome in situ hybridization, but paradoxically it is detected only rarely by routine cytogenetic analysis. Genetic instability, manifesting as a replication error repair phenotype, is found in about 50% of extranodal marginal zone lymphomas. Some cases demonstrate loss of function of A20 , a negative regulator of the NFκB pathway, by mutation, deletion, or promoter methylation.
Reactive lymphoid hyperplasia, including exaggerated Peyer patches.
Other small B-cell lymphomas. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” later in the chapter.
DLBCLs are common in the various extranodal sites. Only those cases with an identifiable low-grade component (extranodal marginal zone lymphoma) can be considered to be related to the group of MALT lymphomas. Even so, use of the term high-grade MALT lymphoma is discouraged.
The importance of recognition of large cell transformation lies in the worsened prognosis (5-year survival for gastric extranodal marginal zone lymphomas dropping from >90% to 60%–70%). However, there is no consensus on the minimum criteria for large cell transformation. When there are compact clusters, confluent aggregates, or sheets of large nucleolated cells outside the confines of the colonized follicles, a diagnostic label DLBCL, arising in extranodal marginal zone lymphoma can be made ( Fig. 21A.84A ). Short of that, the presence of more than 5% interspersed large nucleolated cells merits designation as extranodal marginal zone lymphoma with increased large cells because the prognosis appears to be worsened (see Fig. 21A.84B ).
 The low-grade component (left field) merges into a large cell lymphoma in the right field. (B) There is an increased number of large blastic cells still closely intermingled with the small cells, without forming clusters or sheets. This can be called extranodal marginal zone B-cell lymphoma with increased large cells .")
Genetic data on large cell transformation are limited. In a proportion of cases, TP53 gene inactivation (mutation in one allele, and allelic loss in the other) may mediate the transformation.
Extranodal marginal zone lymphomas are uncommon in the pediatric age group, but show clinical and pathologic characteristics similar to those seen in the adult population. This diagnosis has to be made with great caution in children, because it can be mimicked by marginal zone hyperplasia occurring in various extranodal sites (such as tonsil and appendix). The latter condition features broad marginal zones, prominent intraepithelial infiltration by B cells, and even Ig light chain restriction, but molecular analysis shows a polyclonal pattern.
Nodal marginal zone lymphoma is an uncommon primary nodal B-cell neoplasm morphologically resembling lymph nodes involved by marginal zone lymphomas of extranodal or splenic types, but without evidence of extranodal or splenic disease. By definition, if there is an intimately associated component of follicular lymphoma, the neoplasm is classified instead as follicular lymphoma with marginal zone differentiation .
Nodal marginal zone lymphoma is predominantly a disease of adults, with slight female predominance (see Table 21A.9 ). The patients present with lymphadenopathy. Some may have paraproteinemia. High stage disease (stage III or IV) occurs in 38% to 76% of patients, but B symptoms are uncommon. Compared with extranodal marginal zone lymphoma, bone marrow involvement is more common (29%–62% vs 14%), and the prognosis is worse (5-year overall survival 56%–65% vs 81%; 5-year progression-free survival 35%). Relapse is common. That is, the clinical behavior is more akin to other indolent low-grade B-cell lymphomas. In approximately 20% of cases, the neoplasm transforms into DLBCL, which is associated with a worse prognosis. There are conflicting reports on an association with hepatitis C.
There is usually incomplete architectural effacement of the lymph node. The growth pattern can be diffuse, nodular/follicular, interfollicular, or perifollicular. Reactive lymphoid follicles with well-preserved or eroded mantles may be scattered in the background, and follicular colonization can be present.
The cellular infiltrate is often mixed, including small lymphocytes, cells reminiscent of monocytoid B cells, plasmacytoid cells, plasma cells, and large cells. The monocytoid cells possess oval, notched, or indented nuclei with fine chromatin and inconspicuous nucleoli, and a broad rim of clear to pale cytoplasm ( Fig. 21A.85 ). They often form clusters, bands, and sheets in the sinuses, marginal zones, and interfollicular areas ( Fig. 21A.86 ). Aggregates of plasma cells, which may exhibit cytologic atypia, can be found in some cases; these are often proven to be part of the neoplastic process. There can be some admixed neutrophils. Sometimes, wreaths of epithelioid histiocytes surround clusters of lymphoma cells ( Fig. 21A.87 ). There are interspersed large cells, which may even form small aggregates.

 The lymphoma cells possess round nuclei with superficial notches in the nuclear membrane. There is an appreciable amount of pale cytoplasm, resembling monocytoid B cells. (B) The lymphoma cells more resemble centrocyte-like cells (left field), and many plasma cells (neoplastic) are seen in the right field.")

The immunophenotype is identical to that of extranodal marginal zone lymphoma of MALT. The expressed Ig is most commonly IgM, but can be IgM + IgD, IgG, or IgA. IRTA1 is frequently positive. There is no staining for follicular center cell markers, such as CD10, BCL6, HGAL, and LMO2. BCL2 is usually positive, in contrast to reactive monocytoid B cells. About half of the cases express CD38 or MUM1. CD43 coexpression is present in 24% of cases. Some of the large cells can express BCL6.
The clonally rearranged IG genes usually show somatic hypermutation, consistent with a postgerminal center B cell. Trisomy 3, trisomy 18, trisomy 7, t(3; 14)(q27; q32), structural abnormalities of chromosome 1, and loss of genetic material on chromosome 17 are the most common genetic changes. The chromosomal translocations characteristic of extranodal marginal zone lymphoma are not found, suggesting that these two tumor types are biologically distinct. Similar to extranodal marginal zone lymphoma, some cases exhibit loss of function of A20 .
In 17% of cases of nodal marginal zone lymphoma, the morphologic and immunophenotypic features more closely resemble those of splenic rather than extranodal marginal zone lymphoma. The patients are adults who present with lymphadenopathy but no splenomegaly. Most patients have stage I or II disease. Information on the clinical behavior is limited, but the survival is favorable in the short term.
Histologically the lymph node shows complete architectural effacement by nodular or vaguely nodular lymphoid proliferations. Remnants of germinal centers, often lacking mantle cuffs, can be identified in the centers of some of the nodules. The lymphoma cells are small to midsize, with irregular nuclei, absent or small nucleoli, and pale cytoplasm. Occasional large blastic cells are intermingled with them or are concentrated at the periphery of the nodules. The immunophenotype is similar to the usual nodal marginal zone lymphomas, except for consistent expression of IgD.
Hairy cell leukemia. There is cytologic overlap with hairy cell leukemia, but the clinical setting is totally different.
Monocytoid B-cell hyperplasia. A variety of reactive lymphoid hyperplasias may show prominent monocytoid B cells. The monocytoid B cells are confined to the sinuses and perifollicular zone, and there should not be obliteration of the normal nodal architecture. Presence of large confluent sheets of monocytoid B cells favors a diagnosis of lymphoma. Neoplastic monocytoid B cells are more likely to show greater nuclear irregularities, higher mitotic rate, and more nucleolated forms. In difficult cases, demonstration of light chain restriction, BCL2 immunoreactivity, or molecular clonality studies will confirm a diagnosis of lymphoma.
Systemic mastocytosis. Mast cell disease can mimic extranodal marginal zone lymphoma by virtue of the clear cytoplasm and patchy nodal involvement. Presence of sclerosis and eosinophils should raise the possibility of mastocytosis. See Chapter 21B .
Peripheral T-cell lymphoma.
Marginal zone lymphomas are rare in children, but in contrast to adults, nodal marginal zone lymphoma is more common than extranodal marginal zone lymphoma, and there is a marked male predominance (M:F ratio nearly 20 : 1). The lymphoma tends to present as localized lymphadenopathy (stage I disease). There are disrupted follicles reminiscent of progressive transformation of germinal centers in more than half of the cases, but the overall phenotypic and genetic characteristics resemble those seen in adults. The overall prognosis is excellent.
Diffuse large B-cell lymphoma is an aggressive, rapidly growing neoplasm composed of lymphoid cells with a nucleus comparable in size to or larger than that of a reactive histiocyte, or more than twice that of a normal lymphocyte. This is a heterogeneous category. For cases lacking features of the distinct subtypes, the designation DLBCL, not otherwise specified (DLBCL-NOS) is applied .
DLBCL may arise de novo or via transformation from a low-grade lymphoma, such as follicular lymphoma, CLL/SLL, extranodal marginal zone lymphoma, or lymphoplasmacytic lymphoma. Little is known about the etiology of DLBCL. Some cases occur in a setting of autoimmune disease or immunodeficiency; these are often associated with EBV infection. Some extranodal cases can arise in a setting of local chronic inflammation or irritation, such as postmastectomy lymphedema, chronic suppurative inflammation in bone or soft tissues, metallic implants, and long-standing pyothorax.
DLBCL occurs over a wide age range, with slight male predominance (see Table 21A.9 ). It can present as lymphadenopathy or extranodal disease. The tumors are often fast growing. Constitutional symptoms may be present, particularly in patients with high stage disease. Approximately half of the patients have stage I or II disease at presentation.
On staging, marrow involvement occurs in 16% of cases. It is important to ascertain whether the involvement is by large cell lymphoma (concordant) or an occult follicular lymphoma (discordant), because the prognosis of the former is dismal while the latter does not affect the overall survival, except that the disease is more likely to relapse after complete remission, often in the form of follicular lymphoma.
DLBCL is an aggressive neoplasm, which usually proves fatal if untreated, but it is potentially curable with treatment. Traditional treatment with CHOP chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) or variant has yielded prolonged disease-free survival in 40% to 50% of cases. The addition of rituximab (anti-CD20 chimeric antibody) to the chemotherapy regime has dramatically improved the survival rate by about 20%.
DLBCL is characterized by a diffuse destructive infiltrate in lymph nodes or extranodal sites ( Fig. 21A.88 ). Infiltration of the perinodal tissue and blood vessels is common. Some cases are associated with prominent sclerosis ( Fig. 21A.89 ). Necrosis is common, and the occasional occurrence of total infarction may obscure the diagnosis. Some cases may exhibit a starry-sky pattern imparted by reactive histiocytes. Epithelioid cells, histiocytes, plasma cells, and eosinophils can sometimes be found in the background.
 to the neoplasm.")

The lymphoma cells vary considerably in size from case to case and in the same case. Distinction between the centroblastic (large noncleaved cell) variant and immunoblastic variant is not reproducible, and is optional. The nuclei of the neoplastic cells are typically larger than those of reactive histiocytes, but can be of similar size. They are round, indented, or irregularly folded. The chromatin pattern ranges from vesicular to coarsely granular. Small to large basophilic or eosinophilic nucleoli are always present. They are solitary or multiple, and they are membrane bound or centrally located ( Fig. 21A.90 ). There is a moderate to voluminous amount of cytoplasm, which can be amphophilic, basophilic, pale, or clear. In some cases, the cytoplasm is plasmacytoid, with a pale juxtanuclear Golgi zone. There may be detached fragments of basophilic cytoplasm indistinguishable from the plasma cell bodies seen in inflammatory reactions. Multinucleate cells resembling Reed-Sternberg cells or bizarre cells may be present. Mitotic figures are readily found, and atypical forms are not uncommon. Karyorrhexis may be prominent ( Fig. 21A.91 ). It is not possible to reliably distinguish DLBCL from peripheral T-cell lymphoma on morphologic grounds alone.
 The large cells have small membrane-bound nucleoli. This is traditionally called centroblastic or large noncleaved cell lymphoma. (B) Most large cells are immunoblasts, with central nucleoli. However, there are also some admixed cells with features of centroblasts (large noncleaved cells).")

Large B-cell lymphoma can mimic various sarcomas (such as myxofibrosarcoma or myxoid chondrosarcoma) due to the presence of abundant myxoid stroma in which the round, spindle, or stellate lymphoma cells are suspended ( Fig. 21A.92 ).

The presence of an abundant fibrillary matrix with or without rosette formation can lead to mimicry of neural tumors ( Fig. 21A.93 ). Since the matrix is formed by interlocking cell processes, it stains for CD45 and B-lineage markers.
 Note the fibrillary quality of the eosinophilic-staining rosettes. (B) Immunostaining for CD20 is positive on the cell membranes of the lymphoma cells as well as in the fibrillary materials of the rosettes. It is evident that the fibrillary materials are formed by entangled cell processes.")
Lymphoma cells can assume a spindled configuration as a result of passive entrapment in sclerotic stroma or natural growth in the form of spindle cell fascicles ( Fig. 21A.94 ), mimicking sarcoma. However, areas more diagnostic of lymphoma are often present at least focally.

This is characterized by eccentric displacement of the nuclei by a clear vacuole or eosinophilic globule and can potentially be mistaken for signet ring carcinoma.
Lymphoma cells with clear cytoplasm can be prominent in some DLBCLs and may mimic peripheral T–cell lymphoma, germinoma, or carcinoma ( Fig. 21A.95 ).

Ultrastructurally, some large cell lymphomas exhibit long bushy cell processes, reminiscent of mesothelial cells except for the absence of tonofilaments and desmosomes. These lymphomas usually show no distinctive features on routine microscopy, although a sinusoidal pattern of involvement can sometimes be seen; most are of B lineage. CD30 and EMA are negative. An unusual feature is that approximately 50% of cases express CD56.
Some DLBCLs infiltrate the lymph node in a pure sinusoidal pattern, mimicking metastatic carcinoma or melanoma. The cytologic features are, however, typical of those of lymphoma, and the diagnosis can be confirmed readily by immunohistochemical studies. There is overlap with the microvillous variant.
DLBCLs can sometimes involve predominantly the interfollicular region of the lymphatic tissue, mimicking the growth pattern of peripheral T–cell lymphoma.
Nuclear multilobation, characterized by foldings in the nuclear membrane resembling the petals of a flower, is a not uncommon focal phenomenon in DLBCL and can sometimes be prominent ( Fig. 21A.96 ).

Some DLBCLs are composed of large cells with voluminous cytoplasm and large horseshoe-shaped or bizarre nuclei, sometimes with a sinusoidal growth pattern, mimicking anaplastic large cell lymphoma ( Fig. 21A.97 ). TP53 mutation and genetic abnormalities of MYC, BCL2, and BLC6 are frequent. This variant is associated with aggressive behavior.

Sometimes the large neoplastic cells are admixed with plasma cells, which are shown to exhibit the same Ig light chain. The plasma cells may or may not exhibit cytologic atypia ( Fig. 21A.98 ).

B-lineage markers, such as CD20 and PAX5, are generally positive ( Fig. 21A.99 ). However, the immunophenotype can be aberrant in that one or more markers normally expressed by B cells are lost. Some studies suggest that lack of, or reduced, expression of CD20 or CD22 is correlated with poorer survival. The lymphoma cells often express surface and/or cytoplasmic Ig, but some cases are Ig negative.
 The large cells show strong membrane staining for CD20. (Right) CD3 staining highlights the reactive small lymphocytes.")
CD10 is positive in approximately 40% of cases, and BCL6 in about 60% of cases, consistent with tumors of germinal center cell origin. See “Histogenetic Groups Based on Gene Expression Profiles” later in the chapter, which concentrates on the use of immunostaining for delineation of DLBCL subtypes.
BCL2 is positive in approximately 50% of cases and is associated with unfavorable prognosis and resistance to chemotherapy, especially for the activated B-cell–like group. Activation markers such as CD25 and CD30 are sometimes positive. Aberrant CD43 expression is seen in about 20% of cases. Approximately 10% of cases express CD5; some studies suggest that this subgroup is associated with a worse prognosis. MYC is positive in 10% to 30% of cases, but positive cases do not always show MYC rearrangement. The precise incidence of MYC positivity varies between studies and appears highly dependent on the threshold used to call a case positive (typically ≥40% of cells). The mean Ki67 index is 55%, but an index approaching 100% can occur in some cases.
Rare cases can aberrantly express CD3, causing confusion in lineage assignment. Exceptional cases of DLBCL are cyclin D1 positive, but they are often negative for CD5 and SOX11, and lack CCND1 gene translocation.
IG genes are clonally rearranged and show somatic hypermutation.
In 20% to 30% of cases, the BCL2 gene is translocated to the IGH gene, suggesting that such cases are related to follicular lymphoma. There is no difference in overall survival for the groups with and without BCL2 rearrangement, but the former group has an increased likelihood of relapse, reminiscent of the behavior of follicular lymphoma.
The BCL6 gene is rearranged in approximately 35% of DLBCLs. BCL6 gene, located at 3q27, can be reciprocally translocated to a number of partners, including but not limited to IG gene loci (14q32, 2p12, 22q11). It encodes a zinc finger–type transcription factor normally selectively expressed in follicular center cells, suggesting that it is required for germinal center development and sustainment. BCL6 rearrangement does not show definite prognostic significance. Multiple, often biallelic, mutations are found in the 5′ noncoding region of the BCL6 gene in 73% of cases, irrespective of whether the BCL6 gene is rearranged. BCL6 mutations are accumulated during B-cell transition through the germinal center and thus can also be found in other B–cell lymphomas.
MYC gene rearrangement occurs in about 10% of cases. It usually occurs in a setting of complex genetic alterations and is associated with a worse prognosis.
Mutation in MYD88 gene (L265P) is found in about 30% of the activated B-cell–like subgroup of DLBCLs. The mutation occurs predominantly in primary central nervous system DLBCLs (~30%) and primary cutaneous DLBCL of leg type (~70%). It is a gain-of-function driver mutation, which promotes cell survival. Although this mutation is also present in lymphoplasmacytic lymphoma, the presence of this mutation in DLBCL does not imply origin from LPL.
The REL protooncogene, located at 2p14-15, is amplified in approximately 20% of DLBCLs; this occurrence is seen predominantly in extranodal rather than primary nodal disease. The gene amplification is believed to be a progression-associated event. Mutations in TP53 gene are found in about 20% of DLBCLs and have a negative impact on survival.
Aberrant hypermutations targeting multiple loci, including the protooncogenes PIM1 , MYC , RhoH/TTF (ARHH), and PAX5, occur in more than 50% of DLBCLs. The mutations are distributed in the 5′ untranslated or coding sequences, are independent of chromosomal translocations, and share features typical of IG gene V-region–associated somatic hypermutation as observed in normal follicular center cells. They may contribute to lymphomagenesis by favoring chromosomal translocations through generation of DNA double-strand breaks.
Except in certain variants discussed later in the chapter, sporadic DLBCLs uncommonly harbor EBV (<10%).
Gene expression profiling studies have identified three histogenetic groups of DLBCL :
Germinal center B-cell–like (GCB) group (~50% of cases), which shows high expression of germinal center B-cell signature genes. CD10 expression, BCL2 gene rearrangement, or REL amplification occurs exclusively in this group.
Activated B-cell–like (ABC) group (~30% of cases), which expresses genes normally induced during in vitro activation of peripheral blood B lymphocytes. This group is correlated with the presence of BCL6 translocation, PRDM1/BLIMP1 inactivation, and constitutive activation of NFκB due to somatic mutations in genes encoding the NFκB pathway components, such as A20/TNFAIP3 and CARD11 .
Type 3 group, which does not show the gene expression signatures of either of the other two groups. The molecular pathogenesis of this group remains poorly understood.
These three histogenetic groups exhibit differences in prognosis. The overall 5-year survival rates are 60%, 35%, and 39%, respectively. Some studies, however, fail to show cell-of-origin classification to have prognostic significance.
The classification has therapeutic importance; GCB-type DLBCL has a higher rate of response to intensified immunochemotherapy in the upfront setting and to salvage chemotherapy with R-DHAP following relapse, as compared to ABC-type DLBCL. There are also ongoing trials suggesting benefits of adding lenalidomide or ibrutinib to standard immunochemotherapy in the treatment of the ABC group.
Various algorithms using immunostains as surrogate markers for a GCB or non–GCB (ABC) phenotype have been developed. The Hans algorithm, which is most widely used, includes three markers: CD10, BCL6, and MUM1, with CD10+ or CD10-/BCL6+/MUM1- indicating a GCB phenotype, and CD10-/BCL6- or CD10-/BCL6+/MUM1+ indicating a non-GCB phenotype. The Choi algorithm applies additional markers, including GCET1 and FOXP1. The use of immunohistochemistry to assess cell of origin has been controversial; although the original studies demonstrated good correlation with gene expression profiling and clinical outcome, subsequent studies have yielded conflicting results. The inconsistencies may reflect variations in staining protocols between institutions and subjectivity in result interpretation. However, most available data indicate that classification of DLBCL by cell of origin based on immunohistochemistry correlates with prognosis and response to therapy. The 2017 WHO classification recommends cell-of-origin classification for all DLBCLs and stipulates that both gene expression profiling and immunohistochemistry are acceptable methods. Scaled-down versions of gene expression profiling using NanoString Lymph2Cx platform or reverse transcription-multiplex ligation-dependent probe amplification seems to be more robust than immunohistochemistry in assigning cell of origin for DLBCLs.
Clinical factors. The International Prognostic Index can predict the survival of patients with DLBCLs (see Box 21A.5 ). Bulky tumor and high levels of serum β2-microglobulin are unfavorable prognostic factors.
Morphologic features. Although some studies have shown centroblastic lymphoma to be associated with a better prognosis than immunoblastic lymphoma, the significance of this finding is doubtful because of lack of reproducible criteria to distinguish between them. Furthermore, other studies cannot reproduce the results.
Immunophenotypic features. BCL2 expression is one of the strongest independent unfavorable prognostic factors in DLBCL. This feature appears to confer resistance to chemotherapy, which may be reversed to some extent with addition of rituximab (anti-CD20 antibody) to the chemotherapy regime. High level expression of the MYC oncoprotein is associated with poor outcome, irrespective of whether the MYC locus is rearranged. Coexpression of MYC and BCL2 (often defined by >40% and >50% positive cells, respectively), so-called dual-expressor status, is particularly associated with a poor outcome, indepdenent of genetic rearrangements of either locus, although cases with MYC and BCL2/6 rearrangements tend to behave more aggressively than double-expressor DLBCLs lacking genetic rearrangements. CD5 expression is an unfavorable prognostic feature and correlated with high IPI scores and high stage disease. Absence of HLA-DR expression is associated with a poor prognosis, suggesting that loss of tumor immunosurveillance has a devastating effect on clinical outcome.
Proliferative index. A high proliferative rate (such as Ki67 index >60%–80%) is associated with a poor prognosis and early death in DLBCL.
Germinal center cell differentiation. Gene expression studies have shown that the GCB group of DLBCL is associated with a favorable prognosis. Most but not all studies using other parameters to indicate germinal center cell differentiation have similarly shown a favorable prognosis (e.g., high expression of the HGAL gene, which is normally highly expressed in germinal center B cells, high levels of BCL6 mRNA transcripts, and immunoreactivity for CD10 and BCL6).
Microenvironment. A low percentage of reactive CD8+ T cells (<6%) in the background correlates with a reduction in relapse-free survival. The presence of 15% or more activated granzyme B-positive cytotoxic T lymphocytes is strongly associated with a poor progression-free and overall survival time.
Genetic features. MYC gene rearrangement is associated with a poor prognosis, independent of stage and age. Gains involving chromosomal region 3p11-p12 are associated with a poor prognosis, independent of the gene-expression-based survival model. Mutation of TP53 gene is associated with a poor prognosis. Complex gene copy number alterations often specifically include loss of components of the P53 tumor-suppressor signaling pathway and gains in components that promote E2F-mediated cell cycle progression. De novo tumors with numerous gene copy number alterations (genetically complex) are associated with significantly worse overall survival than those with few alterations (genetically simple) when treated with conventional chemotherapy.
Gene expression profile. Four gene expression signatures and the expression of a single gene BMP6 are strongly correlated with clinical outcome: GCB signature (favorable), MHC class II signature (favorable), lymph node signature (favorable), proliferation signature (poor), and BMP6 (poor). A predictor score calculated from these components is strongly correlated with outcome independent of IPI score or histogenetic type of DLBCL. Other studies have similarly shown the gene expression profile (including genes that regulate responses to B-cell receptor signaling, critical serine/threonine phosphorylation pathways, and apoptosis) to be predictive of prognosis. A redox signature (combining a decrease in antioxidant defense enzyme expression with an increase in thioredoxin system function) is associated with a poor prognosis. One study has shown that quantitation of the expression levels of six genes alone (LMO2, BCL6, FN1, CCND2, SCYA3, BCL2) by real-time PCR can be sufficient for predicting the survival of patients with DLBCL. A subset of DLBCLs shows upregulation of genes involved in mitochondrial oxidative phosphorylation and may be uniquely sensitive to therapeutic perturbation of cellular metabolism.
Metastatic carcinoma or melanoma. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Infectious mononucleosis. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Kikuchi lymphadenitis. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Burkitt lymphoma.
Classic HL. See “Practical Issues in Diagnosis of Lymphoproliferative Lesions” at the end of this chapter.
Some types of DLBCL exhibit distinct clinicopathologic features. These are discussed later in the chapter, except mediastinal large B–cell lymphoma, which is covered in Chapter 21C .
Large B-cell lymphoma harboring translocations of the MYC locus, as well as rearrangement of the BCL2 and/or BCL6 loci, are increasingly recognized as a distinct biologic entity characterized by an extremely aggressive clinical course. It has been suggested that the synergistic actions of MYC and BCL2 are responsible for the poor outcome, with BCL2 driving an antiapoptotic signal and MYC driving a proliferative signal. In the revised 4th edition of WHO classification, these cases are taken out of the DLBCL category and placed in a separate category, high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements; the latter diagnostic category also includes cases with a Burkitt lymphoma–like morphology. However, the more colloquial term double-hit lymphoma remains in wide use. The rare lymphomas with translocations involving all three loci ( MYC, BCL2, and BCL6 ) are termed triple-hit lymphomas .
In some cases, MYC translocation is acquired as part of the transformation from a preexisting follicular lymphoma. In contrast to Burkitt lymphoma, the overall tumor karyotype is usually complex.
Double hit lymphomas account for approximately 5% of all cases originally diagnosed as DLBCL. They generally present in older patients with high stage disease. Extranodal and central nervous system involvement are common. Clinical outcome is poor, with frequent resistance to R-CHOP chemotherapy, inferior survival following hematopoietic stem cell transplantation, and short median overall survival (4.5–34 months) compared to conventional DLBCL. While more intensive chemotherapy regimens (e.g., DA-R-EPOCH and hyper-CVAD) appear to prolong survival, limited and conflicting data have precluded establishment of consensus on the optimal management.
There are no histomorphologic features that reliably predict the double or triple hit status, and cases are frequently indistinguishable morphologically from DLBCL, NOS. Currently the only reliable means to detect MYC and BCL2/6 translocations is to perform FISH or karyotypic analysis, although the positive cases are almost invariably of GCB rather than ABC type.
A subset of cases of double or triple hit lymphoma shows morphologic features overlapping between DLBCL and Burkitt lymphoma. Ideally, the morphology (DLBCL-like, DLBCL/BL, BL-like) should be specified in the pathology report. Importantly, all cases with genetic confirmation of double or triple hit status should be placed in this diagnostic category, as morphology does not appear to impact prognosis. Classification as high-grade B cell lymphoma, not otherwise specified is reserved for cases with morphologic overlap between DLBCL and BL, but lacking genetic evidence of double or triple hit status.
Intravascular large B-cell lymphoma is a peculiar variant of DLBCL characterized by almost exclusive intravascular proliferation of neoplastic cells. There are usually no circulating lymphoma cells despite the intravascular location of the neoplasm. Although bone marrow examination is negative, molecular analysis often shows evidence of involvement.
Most patients are middle-aged or elderly (median age 67–70 years), who present with bizarre neurologic symptoms (due to multiple sites of infarction resulting from occlusion of vasculature) or cutaneous lesions, but any organ (such as lung, kidney, adrenal, and liver) can be involved. Fever is common. Most patients have stage IV disease. The disease pursues a rapidly fatal course, and it is not uncommon for the diagnosis to be made only at autopsy. However, complete remissions can be achieved in some patients given appropriate chemotherapy. The 3-year overall survival is 43% to 52%.
Noncohesive large lymphoma cells with vesicular nuclei and distinct nucleoli characteristically plug the lumens of small and midsize blood vessels ( Fig. 21A.100 ). The tumor cells are often enmeshed in fibrin or platelet thrombi. The vascular occlusion often leads to extensive infarcts. The tumor cells may palisade along the luminal side of the blood vessels, simulating endothelial lining. An extravascular component of lymphoma is sometimes seen.

The great majority of cases are of B lineage, although rare cases exhibiting T, NK, or histiocytic lineage have also been reported (with such cases being not classified under the category intravascular large B-cell lymphoma ). A subset of cases expresses CD5. Expression of CD11a and CD49a on the lymphoma cells and expression of the corresponding ligands CD54 and CD106 on the endothelial cells may explain the tendency of the lymphomas to localize within the intravascular compartment. IG genes are clonally rearranged. EBV is usually negative.
Malignant histiocytosis-like large B-cell lymphoma represents the so-called Asian type of intravascular large B–cell lymphoma. The patients present with fever, hepatosplenomegaly, and hemophagocytic syndrome without lymphadenopathy or skin lesions. The disease pursues an aggressive clinical course. Bone marrow biopsy shows sinusoidal infiltration by neoplastic large B cells, accompanied by hemophagocytic histiocytes. The liver shows sinusoidal and portal infiltration by neoplastic cells and hemophagocytic histiocytes. The infiltrate in the spleen occurs in the red pulp, without tumor nodule formation.
THRLBCL is a large B-cell lymphoma accompanied by a prominent component of reactive T lymphocytes and frequently histiocytes, with these reactive cells constituting more than 90% of the total cell population. It is controversial whether THRLBCL represents a distinctive clinicopathologic or merely a morphologic variant of DLBCL. Some cases appear to represent a progressed form of follicular center cell lymphoma or NLPHL.
The inclusion criteria in the reported series vary widely, and thus the findings are not always easy to interpret and compare. Some studies report that THRLBCL behaves no differently from conventional DLBCL. However, using strict criteria for diagnosis, more than 90% of patients have stage III or IV disease, and bone marrow involvement is common (>50%). The prognosis is poor, with 5-year survival of only 20% ; however, the poor prognosis may be explainable by the high proportion of patients having high stage disease and high IPI.
THRLBCL is characterized by isolated or small groups of large lymphoma cells dispersed in a background of small lymphocytes, which may be admixed with variable numbers of histiocytes, epithelioid histiocytes, eosinophils, and plasma cells ( Fig. 21A.101 ). The large cells have round, lobated, or irregularly folded nuclei and distinct nucleoli, and can resemble L&H cells or Reed-Sternberg cells. By definition, they should not occur in dense clusters or sheets. The background small lymphocytes have small dark round nuclei, or appear mildly atypical, with irregular or elongated nuclei. A case would be excluded from this category if there is any identifiable component of NLPHL (i.e., the diagnosis would have been NLPHL with diffuse component ).
 Atypical large cells are sparsely scattered in a background of small lymphocytes. (B) In this example, the large cells exhibit irregular nuclear foldings. Many histiocytes are also admixed, conforming to so-called T-cell/histiocyte-rich large B-cell lymphoma. Distinction from nodular lymphocyte predominant Hodgkin lymphoma is difficult.")
B-cell markers will highlight the individually dispersed large cells, and light chain restriction can sometimes be demonstrated ( Fig. 21A.102 ). There should be few small B cells in the background. BCL6 is frequently expressed. Consistent expression of EMA has been reported in some but not all series. Occasional cases can be CD30+. The background contains numerous small T lymphocytes that often express CD8 and cytotoxic markers (see Fig. 21A.102 ). There are no FDC meshworks in the background. PD-L1 is frequently overexpressed.
 The large neoplastic cells are strongly and uniformly highlighted by CD20 staining. They do not form large clusters or sheets. (Right) Many small T cells (CD3+) are present in the background.")
Molecular studies show clonal rearrangements of IG genes. EBV, with rare exceptions, is negative.
Many differential diagnoses have to be considered, such as reactive immunoblastic proliferation, CHL, NLPHL with diffuse component, and peripheral T–cell lymphoma.
Lymphomatoid granulomatosis was originally described by Liebow et al as a pulmonary angiocentric polymorphous atypical lymphoreticular infiltrate accompanied by prominent necrosis. It was uncertain as to whether it was a neoplastic or inflammatory process. Subsequently the disease was recognized also to affect the skin, central nervous system, and kidneys. In the late 1970s and early 1980s, similarities between lymphomatoid granulomatosis of the lung and polymorphic reticulosis of the nasal cavity were noted. Jaffe introduced a unifying concept of angiocentric immunoproliferative lesions to encompass these lesions, and they were thought to represent a distinctive form of T-cell lymphoma since large numbers of T cells are demonstrated by immunohistochemistry. Reassessment of the entity, however, shows that while polymorphic reticulosis of the nasal cavity is usually an NK/T-cell lymphoma, lymphomatoid granulomatosis is an EBV+ clonal B-cell proliferation associated with a prominent reactive T-cell component.
The median age is in the fifth to sixth decades, and there is male predominance. Most patients present with pulmonary lesions, manifesting as cough, chest pain, or dyspnea. Chest x-ray usually shows bilateral peripheral lung nodules, solitary mass, or diffuse infiltrates. Others present with extrapulmonary disease, such as in the skin (macular rash, dermal nodules, or plaques), central nervous system (mass lesion or cortical infarct, causing confusion, dementia, ataxia, or cranial nerve palsy), kidney, and other organs. The bone marrow is usually not involved. The tumor sometimes occurs in a setting of immunodeficiency, including AIDS and posttransplant. Even in the absence of overt immunodeficiency, the patients are often found to have defective cell-mediated immunity.
The histologic triad is (1) atypical lymphoid infiltrate, which is often mixed; (2) vascular invasion (angioinvasion); and (3) prominent coagulative necrosis, which is often geographic ( Fig. 21A.103 ). The cellular infiltrate often includes small lymphocytes, large atypical lymphoid cells, and histiocytes, sometimes with granuloma formation. The large atypical lymphoid cells can resemble centroblasts, immunoblasts, or Reed-Sternberg cells. Not uncommonly they are bizarre, with huge and irregularly folded nuclei and coarse chromatin ( Figs. 21A.104 and 21A.105 ). The large atypical cells range from scanty (sometimes requiring immunostaining to highlight them) to numerous, and a histologic grade can be assigned according to their number, from grade I (few atypical large cells) through grade II (obvious mixture of small and large lymphoid cells) to grade III (many large atypical cells).
.")

 Many large atypical cells are present, and the cell composition is similar to that of large cell lymphoma. (Right upper) CD20 immunostaining highlights large number of neoplastic cells (B lineage) infiltrating the blood vessel wall. (Right lower) The atypical large cells are highlighted by in situ hybridization for EBER.")
CD20 staining selectively highlights the large atypical cells, many of which are proliferating, as evidenced by Ki67 staining (see Fig. 21A.105 ). The CD20+ large cells are usually negative for CD30 and CD15. The background is rich in reactive small T lymphocytes, which are mostly cytotoxic CD8+ cells. The B-cell nature of the disease process is further confirmed by the presence of clonal IG gene rearrangements. EBV can be demonstrated in the large atypical cells (see Fig. 21A.105 ).
The natural history is highly variable. Spontaneous regression can occur in 14% to 27% of patients, but most show disease progression, resulting in death if treatment fails. Adverse prognostic factors are histologic grade II or III, age over 25 years, and presence of neurologic disease. Lower grade lymphomatoid granulomatosis occasionally undergoes spontaneous remission and is best managed with strategies designed to enhance the host's underlying immune system (such as interferon α2b), whereas high-grade disease is best managed by combination chemoimmunotherapy, but this has inferior outcomes.
Although lymphomatoid granulomatosis can be considered conceptually as a form of THRLBCL, it shows a number of distinctive features that set it apart:
Exclusive extranodal location (the disease is either nodal or extranodal in THRLBCL)
Coagulative necrosis being a constant feature (absent or focal in THRLBCL)
Angiocentric-angiodestructive growth as a prominent feature (usually absent in THRLBCL)
Consistent association with EBV (usually negative in THRLBCL)
EBV+ DLBCL, NOS is an EBV-associated DLBCL occurring in a host without evidence of overt immunodeficiency and not conforming to other defined entities, such as lymphomatoid granulomatosis, DLBCL associated with chronic inflammation, and plasmablastic lymphoma. This nomenclature replaces the provisional category EBV+ DLBCL of the elderly in the 2008 WHO classification, with age restriction removed because EBV+ DLBCLs can also occur in younger patients. The disease may involve nodal or extranodal sites (especially lung and gastrointestinal tract). Constitutional symptoms such as fever and weight loss can be present. In elderly patients, the lymphoma is aggressive, with median survival of 2 years. In contrast, in young patients, the outcome is excellent.
Histologically there is often coagulative necrosis and angiocentric-angiodestructive growth. The lymphoid infiltrate can be either polymorphic (with large lymphoid cells admixed with small lymphocytes and plasma cells, resembling T-cell/histiocytes-rich large B-cell lymphoma or classical Hodgkin lymphoma) or monomorphic (like conventional large B-cell lymphoma). Some of the large cells are often giant and bizarre, and may resemble Reed-Sternberg cells. The immunophenotype of the large cells is CD20+, CD79a+, CD30+-, CD5-, CD10-, EBV LMP1+, EBNA2-/+.
EBV+ mucocutaneous ulcer is a distinct form of EBV+ DLBCL that occurs on cutaneous or mucosal surfaces in the setting of age-related or iatrogenic immunosuppression. It typically presents as a well-circumscribed ulcerative lesion involving the oral or gastrointestinal mucosa, and is comprised of large cells, some of which resemble Reed-Sternberg cells. Immunophenotypically, the cells are nearly always positive for CD20 and CD30, and may be CD15+ as well. There are many admixed small T lymphocytes, especially at the base of the lesion. The lesion tends to be self-limited and resolves with conservative management or reduction of immunosuppressant.
First described among patients with long-standing pyothorax, the tumor involves body cavities or confined body spaces (e.g., joint space, space around prosthesis) in the setting of chronic inflammation. The interval between the initiating event (such as the creation of an artificial pneumothorax or implantation of prosthesis) and the presentation of lymphoma is years to decades. The disease is aggressive, with a median survival of 5 to 9 months. Pyothorax-associated DLBCL, the prototypic type of DLBCL associated with chronic inflammation, complicates long-standing pyothorax resulting from artificial pneumothorax for the treatment of pulmonary tuberculosis or tuberculous pleuritis. It is a pleura-based mass-forming tumor, which is in contrast to the lack of mass lesion in primary effusion lymphoma ( Fig. 21A.106A ). The mean interval between the history of chronic pyothorax and diagnosis of lymphoma is 37 years, and it has been suggested that chronic inflammatory stimulation of nonautoimmune nature may be the etiologic factor. Originally reported in Japanese, this lymphoma type is also recognized in other Asians and Caucasians. The patients are adults with a median age of 64 years and marked male predominance (M:F >10 : 1). The commonest presenting symptoms are chest pain, productive cough, dyspnea, and chest wall mass. By the time of diagnosis there is usually invasion of the chest wall, lung, pericardium, and diaphragm, but distant spread is uncommon.
 CT scan shows tumor mass in the pleura with invasion of the chest wall. Pleural effusion is also present. (B) The lymphoma is composed of large cells with moderate amount of cytoplasm. These cells are positive for EBNA2 (inset).")
There are also rare cases found incidentally (such as in splenic cyst, adrenal pseudocyst, renal pseudocyst, hydrocele, arachnoid cyst, chronic hematoma, cardiac myxoma, thrombus, endovascular graft, or around metallic prosthesis) in the absence of a frank tumor mass. These cases show excellent outcome with surgical excision alone and/or chemotherapy; it is recommended that these cases be termed fibrin-associated DLBCL to emphasize the marked difference in clinical outcome.
For pyothorax-associated DLBCL, the pleura shows marked fibrous thickening and dense infiltration by a large cell lymphoma (see Fig. 21A.106B ). For fibrin-associated DLBCL, large lymphoma cells are present in low numbers and often occur in aggregates within a background of abundant fibrinous or proteinaceous material.
The neoplastic cells are generally positive for B-cell antigens such as CD20 and CD79a . However, because the tumor is of a nongerminal center B-cell type, there may be downregulation or loss of CD20, and upregulation of MUM1 and CD138. Immunoglobulin light chains are monotypic, although, remarkably, coexpression of T-cell antigens (CD2, CD3, or CD4) can be seen in some cases.
The tumor is invariably positive for EBV. Like immunodeficiency-related EBV+ tumors, the EBV demonstrates a type III latency program (i.e., EBNA2+ in addition to LMP1+), suggesting that the lymphoma has developed within a milieu of localized immunodeficiency (see Fig. 21A.106B ). The lymphoma is negative for HHV8.
At the genetic level, TP53 gene mutation is very common (70%). Pyothorax-associated DLBCL shows a gene expression profile that differs from that of conventional DLBCL; in particular, there is striking overexpression of interferon-inducible protein 27.
Primary effusion lymphoma is a form of DLBCL of plasmablastic phenotype, occurring almost exclusively in patients with AIDS. The patients present with pleural, pericardial, or peritoneal effusion, without formation of a discrete tumor mass. They have a median age of 41 years with marked male predilection (M:F ratio 8.5 : 1). The tumor is highly aggressive. The median survival is 6.2 months, and the 1-year overall survival rate is only 30% to 39%. An extracavitary solid form of the lymphoma involving lymph nodes or various extranodal sites, without an accompanying effusion, is also recognized. The main differences from classic primary effusion lymphoma include a slightly higher frequency of expression of B-cell antigens (25%) and Ig (25%), and a better survival (median 11 months vs 3 months).
The neoplastic cells are large, with voluminous basophilic cytoplasm and a pale Golgi zone. The nuclei are often highly pleomorphic ( Fig. 21A.107 ). In the extracavitary form, the lymphomatous infiltrate is diffuse, often accompanied by high mitotic activity and apoptotic debris.
. There are huge cells with pleomorphic nuclei and abundant basophilic cytoplasm.")
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here