Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

Previously known as congenital cystic adenomatoid malformation (CCAM)
A rare lesion caused by hamartomatous proliferation of the terminal bronchioles at the expense of alveolar development ▸ it is characterized by a multicystic mass of pulmonary tissue with proliferation of the bronchiolar structures
It is usually unilobar and communicates with a normal tracheobronchial tree ▸ it receives its blood supply from a normal pulmonary artery and vein ▸ it may compress the contralateral lung (resulting in hypoplasia)
Type 1 – macrocystic (65%): variable cysts with at least one dominant cyst present (up to 10 cm) ▸ this has a good prognosis (and an infrequent association with other congenital abnormalities)
Type 2 – microcystic (10–15%) : smaller more uniform cysts (up to 2 cm) ▸ congenital malformations are common (50%)
Type 3 – mixed (~8%) : solid-appearing microcysts (1.5 cm) with an associated mass effect (giving a ground-glass appearance on CT) ▸ it has a poor prognosis due to the associated congenital malformations and severe respiratory compromise ▸ nearly always mediastinal shift
Type 4 (10–15%): large cysts (up to 7 cm) ▸ may be a precursor of pleuropulmonary blastoma
Neonatal respiratory distress or it can present in older children ▸ symptoms are due to a combination of obstructive emphysema, mediastinal shift and infection
During the first few hours of life a CCAM will appear as a soft tissue mass (due to the retained lung fluid within it) which may cause mediastinal shift ▸ once this fluid has been reabsorbed it will be replaced by an air-filled cystic lesion
This can distinguish between a CCAM and a congenital diaphragmatic hernia
Echogenic soft tissue mass ▸ multiple variable size anechoic cysts or a homogeneous echogenic solid mass ▸ large lesions can cause mediastinal shift (± lung hypoplasia)
T2WI: hyperintense ▸ uni-/multilocular or solid ▸ over time the fluid can be replaced by air (± air–fluid levels)
CPAMs can communicate with the airways ▸ infected (30%) ▸ blood supply from the pulmonary artery with drainage via pulmonary veins
There is a variable natural history and prognosis (larger lesions have a worse prognosis)
Lesions that are identified prenatally may involute in utero
Treatment: early surgical removal may be required if there is severe pulmonary compromise (and if the contralateral lung is not hypoplastic)
 CXR shows a large air-filled abnormality in the left lung causing marked contralateral mediastinal shift. Attempts have been made to insert intercostal drains. (B) Coronal CT reformat confirms the presence of a large multicystic mass. Note the narrowed and displaced left main bronchus. **")
 CXR demonstrating extensive ground-glass opacification with gross overinflation of the right lung and herniation across the midline. (B) CT again demonstrates overexpansion of the right lung with ground-glass shadowing due to microcysts beyond CT resolution. †")
This anomaly shares some features with sequestration except:
The lung is normally connected to the bronchial tree
The vein draining the affected lobe (usually the right lower lobe) drains into the IVC or portal vein (rather than the left atrium)
There is usually an absent or small pulmonary artery perfusing the abnormal lung
The arterial supply is partly or wholly from the thoracic or abdominal aorta, or the coeliac axis
It may be asymptomatic or present with features of a left-to-right shunt
A small ipsilateral lung with ipsilateral mediastinal shift
The abnormal pulmonary vein is seen draining down and enlarging towards the diaphragm in the shape of a ‘scimitar’ sword
. (A) CXR showing (1) shift of the heart into the right hemithorax, (2) a small right lung with an abnormal vessel (arrow) paralleling the right heart border and (3) overinflation (compensatory) of left lung. (B) Coronal CT reformat highlights the abnormal ‘scimitar vein’ (arrow) draining below the diaphragm into the systemic venous system bypassing the pulmonary veins. **")
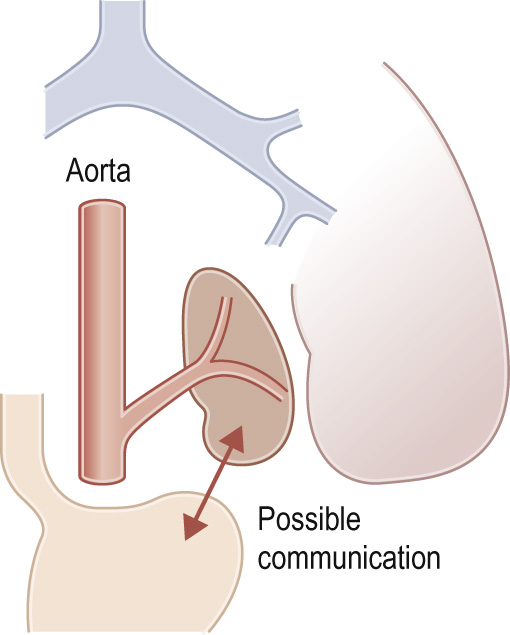
A congenital mass of aberrant pulmonary tissue that has no normal connection with the bronchial tree or pulmonary arteries ▸ lesions are defined as either intra- or extralobar
It derives its arterial supply from either the thoracic or abdominal aorta ▸ its venous drainage can either be via the pulmonary or systemic veins
It can present at birth or in an older child (depending on the type) with recurrent focal infections, bronchiectasis, haemoptysis or as an asymptomatic pulmonary mass
A solid well-defined highly echogenic mass ▸ the anomalous systemic arterial supply is difficult to visualize despite the availability of colour flow Doppler
A persistent multicystic basal opacity which is frequently left sided
This enables definition of the arterial and venous vascular anatomy
Solid, well-defined hyperintense T2WI mass
Extralobar sequestration can be located below the diaphragm and mimic a neuroblastoma or adrenal haemorrhage
 Axial CECT through the lung bases with a large systemic vessel arising from the left side of the aorta (arrow A) supplying a very vascular left-sided extralobar sequestration (arrow B). (B) Coronal CT showing the normal lung and beneath this (arrow) the left-sided basal extralobar sequestration with a draining vein entering the azygous system below the diaphragm. †")
. (A) PA CXR. The pulmonary vessels at the right base display an abnormal course ▸ this suggests they may be draped around a space-occupying but air-filled lesion. The right hemidiaphragm is slightly depressed and the heart is shifted slightly to the left. (B) Aortogram demonstrates a large single vessel arising from the distal aorta supplying a portion of the right lower lobe. (C) CECT confirms the vascular supply.")
 Axial CT shows an enhancing mass in the posterior left lower lobe with a large (enhancing) feeding vessel (arrow). (B) Oblique coronal reformat highlights the mass receiving arterial supply from a branch of the coeliac artery (arrow) with venous drainage occurring via a left pulmonary vein (arrowhead). **")
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here