Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124


Acanthosis nigricans is a commonly encountered skin dermatosis that can be seen in various clinical scenarios. It is overwhelmingly associated with obesity but can occur secondary to medications, endocrine disorders such as the HAIR-AN syndrome ( h yper a ndrogenism, i nsulin r esistance, and a canthosis n igricans), diabetes, and internal malignancies. This last type is clinically distinctive and manifests in a unique manner.
Clinical Findings: Classic cases of acanthosis nigricans affect the nape of the neck, the axillae, and the groin regions. Native Americans and African Americans are at a significantly increased risk for development of acanthosis nigricans. The slow, insidious onset of patches and plaques with a velvety, hyperpigmented, thickened, rough surface is characteristic of acanthosis nigricans. Maceration with a malodorous smell is often noted. The patients are for the most part asymptomatic, although some complain of intermittent pruritus. The clinical findings in association with obesity are enough to make the diagnosis. A thorough history should be taken to rule out a medication-induced form of acanthosis nigricans. The only routine laboratory testing performed is screening for occult diabetes. Patients with obesity are at higher risk for diabetes later in life, and lifelong follow-up and screening by their primary care physician is required.
Many medications have been shown to induce acanthosis nigricans. They include niacinamide, glucocorticoids, insulin, and some birth control pills. The medication most commonly associated with acanthosis nigricans is niacinamide. Most cases resolve or improve greatly with discontinuation of the medication. The appearance is often identical to that of classic acanthosis nigricans, but the history is suggestive, with the timing of rash onset related to the introduction of the causative medication.
Malignancy-associated acanthosis nigricans is often widespread and involves unique areas, including the mucous membranes, palms, and soles. This form has a rapid onset and affects different areas of the body than the classic form of acanthosis nigricans does. The palms and soles are often involved, and the face can be involved. Any case in which there is rapid onset of acanthosis nigricans in a widespread distribution, often in a nonobese individual, warrants proper evaluation to rule out an internal malignancy. Referral to a gastroenterologist and an internist for cancer screening is of utmost importance.
A few endocrine disorders can be associated with acanthosis nigricans, most frequently diabetes mellitus and the HAIR-AN syndrome It is associated with insulin resistance and also with hyperandrogenism.
Rare causes of acanthosis nigricans include the familial forms, which are inherited in an autosomal dominant fashion.
Pathogenesis: The skin thickening and clinical findings are possibly caused by an increase in insulin-like growth factor receptor, fibroblast growth factor receptor, and epidermal growth factor receptor and their subsequent effects on the skin. The reason it affects certain regions preferentially is unknown. Malignancy-associated acanthosis nigricans is believed to be caused by some cytokine or growth factor directly secreted by the tumor, possibly in the fibroblast growth factor receptor class of molecules. The tumor causes the clinical findings by secreting these substances. Acanthosis nigricans is believed to be a paraneoplastic process in these cases. Medication-induced acanthosis nigricans is poorly understood but is possibly related to the medication's local effects on the skin in genetically predisposed individuals.
Histology: Epidermal hyperplasia, acanthosis, and papillomatosis are present. There is minimal to no inflammatory infiltrate, and the dermis is essentially normal in appearance. Extensive hyperkeratosis with a mild excess of melanin production likely explains the hyperpigmentation seen in acanthosis nigricans.
Treatment: Treatment is often difficult unless the afflicted individual makes a conscious effort to get to an ideal body weight and to get his or her diabetes under excellent control. This is the only likely scenario in which the skin findings of acanthosis nigricans will resolve. Temporizing methods of therapy include the use of keratolytic agents such as lactic acid to help thin the plaques and make them less noticeable. These agents are difficult to use in the axillae because of stinging. The topical use of tretinoin cream has also been successful. Destructive laser therapies have been used with varying success.
Treatment of malignancy-associated acanthosis nigricans is directed at the underlying malignancy. Removal of the tumor may result in complete resolution of the skin disease.
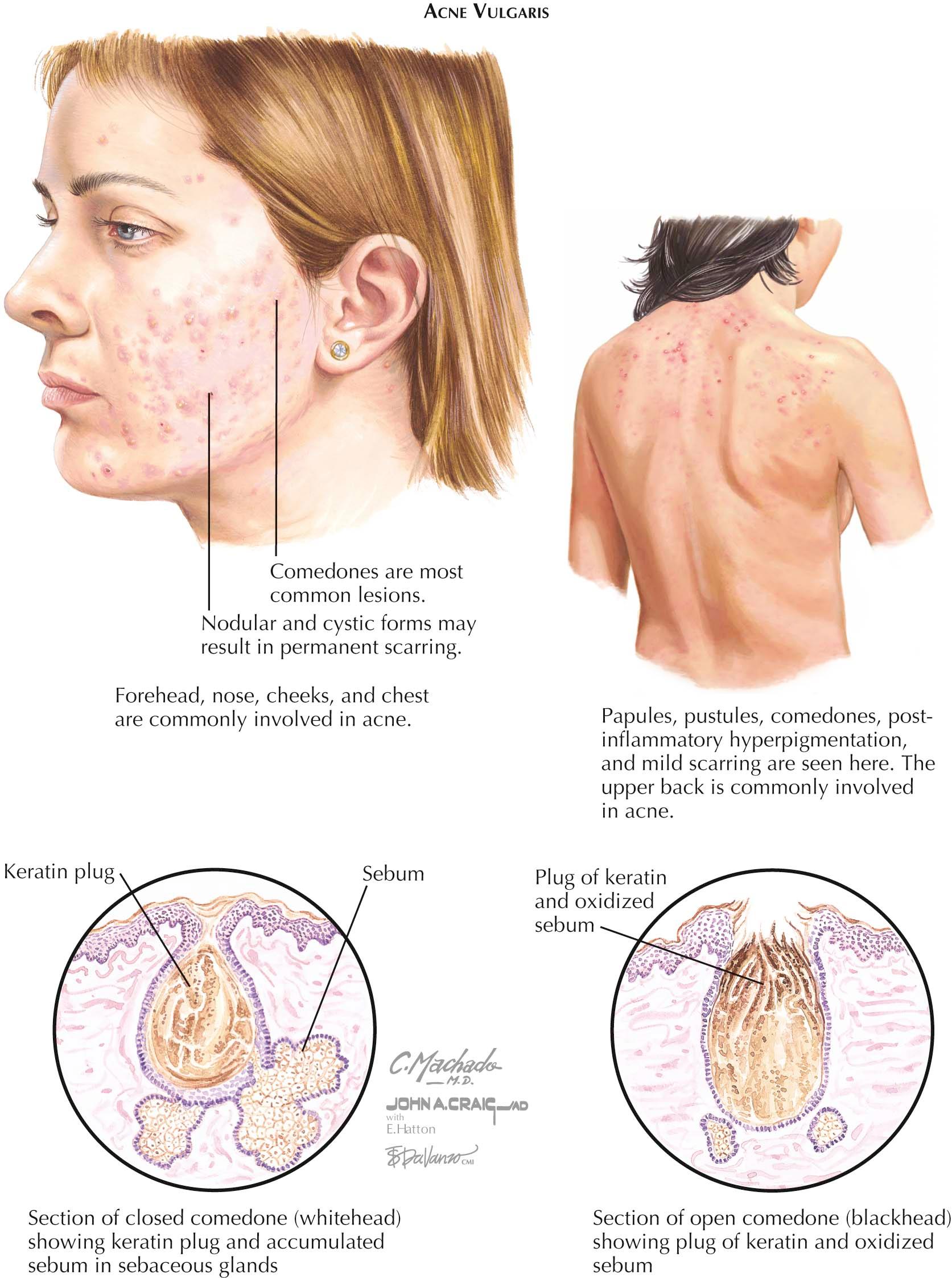

Acne is an almost universal finding in teenagers across the globe. Acne vulgaris is the most common form of acne; it affects almost every human at some point in their lifetime. Most cases are mild and do not cause any significant disease. Most acne vulgaris is seen in the postpubertal years. Many clinical variants exist, and excellent therapeutic modalities are available to treat this skin disease.
Clinical Findings: Acne vulgaris typically begins soon after puberty. It has no racial or gender preference, although males may develop more severe cases of the disease. The first signs of acne development are the formation of microcomedones, both open and closed. Open comedones, also known as blackheads, appear as small (0.5-1 mm), dilated skin pores that are filled with a dark material, oxidized keratin. This material can be easily expressed with lateral pressure or with the help of a comedone extractor. The closed comedone, also known as a whitehead, is a small (0.5-1 mm), whitish to skin-colored papule. Comedones are believed to be the precursor lesion to the other lesions of acne. As acne progresses, inflammatory red, slightly tender papules develop, along with a variable amount of pustules. The pustules are centered on the hair follicle. More severe cases of acne, such as nodulocystic acne, show inflammatory nodule formation as well as cyst formation. These nodules and cysts can become large (2-3 cm in diameter) and can cause considerable pain. They often heal with scarring of the skin.
The face, back, upper chest, and shoulders are the predominant areas of involvement, most likely because of the higher density of sebaceous glands in these regions and the role of the sebaceous gland in the development of acne. Acne is a relentless condition: As one lesion heals, another develops simultaneously. Females often report a flare of their acne 1 week before menstruation begins, denoting hormonal influence. Acne has many clinical variants.
Adult female acne is typically seen in women between 25 and 45 years of age. They often report that they had minimal to no acne during adolescence. This form of acne is found predominantly on the cheeks, perioral region, and jaw line, and it manifests as deep-seated papules, nodules, and cysts. There is a pronounced flare around the time of menstruation.
Neonatal and infantile acne are self-limited types that are seen frequently in this population. Neonatal acne may be seen a day or two after delivery; it is caused by transplacental passage of maternal hormones. It resolves without therapy and seems to be more prevalent in male newborns. Infantile acne is seen after the first few months of life. Most cases show a few transient papules, comedones, and pustules. Most self-resolve, although a few cases last into adolescence.
Acne cosmetica and acne medicamentosa are two similar forms of acne thought to be caused by or exacerbated by the use of cosmetics and facial medications. The removal of these products usually is enough for the patient to see significant improvement. Most products implicated in this form of acne are oily in nature; they cause follicular plugging, which allows acne production.
Acne excoriée is a form of acne that is made worse by chronic picking or manipulation of the acneiform lesions. This often leads to scarring and a worsening of the clinical appearance. It is often coexistent with an underlying anxiety disorder, obsessive compulsive disorder, or depression.
Rare forms of acne include acne fulminans, acne conglobata, and acne aestivalis. Acne fulminans is seen almost exclusively in teenage boys. It is a form of severe cystic nodular acne that heals with severe, disfiguring scarring. The cysts and nodules can easily rupture and break down, leaving multiple ulcerations. This is associated with systemic symptoms including fever, arthralgias and arthritis, and myalgias. A peripheral leukocytosis is often seen on laboratory examination. Lytic bone lesions can be seen, with the clavicle the most commonly affected bone. This may be preceded by localized pain over the bony involvement. Acne conglobata is a term used to refer to severe cystic acne, which is seen mostly in young males. Patients often have multiple cysts that can be interconnected with sinus tracts. The areas involved are very painful and heal with severe scarring. This form of acne occurs in the same locations as acne vulgaris. Acne conglobata has been seen in association with hidradenitis suppurativa, and some consider these conditions to be in the same spectrum of disease processes. Acne conglobata may run a chronic course well into adulthood, with persistent nodules and cysts coming and going. Acne aestivalis is one of the rarest forms of acne. It has a seasonal variation to its course. It begins in spring and resolves by early fall. It is a disease predominantly of adult women.
Steroid-induced acne occur secondary to the chronic use of oral or intravenous steroids. It manifests as a monomorphic eruption of inflammatory papules. Many other medications can be associated with acneiform eruptions, including iodides, lithium, and the epidermal growth factor inhibitors.
Pathogenesis: Acne is believed to have a multifactorial basis. Follicular keratinization appears to be faulty, and the keratinocyte adhesions do not separate as quickly as they should, leading to a follicular plug and microcomedone formation. Excessive sebaceous gland production also plays a role and is probably mediated by hormonal influences. If the sebaceous gland material is produced in an amount sufficient to cause rupture of the comedone, the contents spill into the dermis, causing an inflammatory response; clinically, this is manifested by inflammatory papules, nodules, and cysts. The third player in the pathogenesis is the gram-negative anaerobic bacteria, Propionibacterium acnes . This bacteria is believed to cause an activation of the immune system and results in an inflammatory infiltrate. Rare causes of acne include adrenal gland disorders that can cause virilization. These tumors are rare and often are associated with a sudden onset of acne, hirsutism, and irregular menstrual cycles. Any state of hyperandrogenism can cause acne or make preexisting acne worse. The most common cause is the polycystic ovarian syndrome in women. Less commonly, a Sertoli-Leydig cell tumor can lead to a hyperandrogenic state and resultant acne.
Histology: Biopsies of acne are not required for diagnosis. A biopsy specimen from an inflammatory acne papule shows a folliculocentric lesion with a dense inflammatory infiltrate. The follicular epithelium has signs of spongiosis. Foreign body giant cells, plasma cells, neutrophils, and lymphocytes are all seen in varying degrees. Comedones show compacted corneocytes within the sebaceous gland lumen.
Treatment: Treatment for acne vulgaris is multidimensional. One often uses a combination of a keratolytic and antibacterial agent, such as benzyl peroxide, with tretinoin (a medication that increases differentiation and maturation of keratinocytes) and an antibiotic. The antibiotics are used for their antiinflammatory and antibacterial properties. The antibiotic may be given in a topical or oral form. More severe acne, cystic acne, acne conglobata, and acne fulminans require the systemic use of isotretinoin to prevent severe scarring. Isotretinoin is given for 5 to 6 months. Significant precautions need to be taken, because this medication is a well-known teratogen. Prednisone is often advocated for these severe cases of cystic acne. It is usually used transiently, when first beginning therapy with isotretinoin, to help decrease some of the severe inflammation. It should not be used for long periods.
Many other treatment options exist, including topical agents such as azelaic acid, adapalene, tazarotene, salicylic acid, and topical antibiotics. Oral medications that can be used include multiple oral antibiotics, spironolactone, and birth control pills. The latter two medications are especially helpful in the treatment of adult female acne. They work on the hormonal influence on acne and are highly successful in this type of patient. All the medications used for acne have potential side effects, and treatment must be tailored to the individual. Comedone extraction, intralesional triamcinolone, and photodynamic therapy have shown some success in treating acne. Laser resurfacing, chemical peels, and use of artificial fillers should be reserved for the treatment of scarring after the inflammatory acne has been controlled.

Acne keloidalis nuchae is a fairly common form of inflammatory, scarring alopecia that typically occurs on the posterior occipital scalp. There is a variable spectrum of disease, ranging from very mild cases to severe scarring alopecia. The condition has psychosocial implications and is difficult to treat effectively. It is diagnosed clinically, and biopsies are rarely needed.
Clinical Findings: Acne keloidalis nuchae begins on the posterior scalp or nape of the neck as tiny, follicular, flesh-colored to red papules. The papules enlarge to form plaques, which coalesce into larger plaques. Ultimately in severe cases, the entire posterior scalp is involved. Early in the disease, no hair loss is appreciated. As the disease progresses, the hair follicles become scarred down and crowded out by the encroaching fibrosis, resulting in a variable amount of scarring alopecia.
This condition is far more common in young adult men, with a predilection for African Americans. It was originally believed to be caused by close shaving of the hair and the subsequent inflammation caused by the newly regrowing hair as it pierces the epidermis. The curly nature of the hair follicle was believed to be one of the most important factors. This theory of the pathophysiology of the disease has been questioned, and the cause of the condition is not as simple as once theorized.
The plaques, if left untreated, eventually form thickened scar tissue resembling the appearance of a keloid scar. The scarring alopecia is permanent, and the patient is left with a considerable cosmetic issue. Severe cases of this condition can cause psychological issues, as can almost any form of severe alopecia.
Pathogenesis: Originally, acne keloidalis was believed to be caused by the close haircut in African American men, which caused the hairs to penetrate the epidermis on regrowth, setting off an inflammatory reaction. It has now been determined that this is an oversimplification of the disease state. Other factors are likely to play more important roles in the pathogenesis.
Histology: Early disease often appears as a dense, mixed inflammatory infiltrate around the hair follicle and adnexal structures with plasma cells present. This appears similar to folliculitis. As the hair follicles rupture, the contents spill into the dermis and set off a dermal inflammatory reaction. There is overlying epidermal hyperplasia and acanthosis. Occasional pustule formation is seen and is composed of pools of neutrophils.
Late disease is very similar to the pathology of a keloid. There is a lack of adnexal structures and fibrosis throughout the dermis.
Treatment: Therapy for mild disease requires a multifaceted approach. If only a few papules are present with minimal hair loss, a combination of a topical and an oral antibiotic can be used for their antiinflammatory effects. The most commonly used oral antibiotics are in the tetracycline class. The topical antibiotic most often prescribed is clindamycin. Strict hair care regimens are required to help decrease the trauma to the skin. Shaving of the scalp should be avoided, and haircuts with shears should also be minimized, because the shears can cause microtrauma to the skin and potentially induce the process and scarring formation. Cutting the hair to a length of 3 to 5 mm is a reasonable approach that minimizes trauma to the skin. Topical retinoids such as tretinoin and tazarotene have been used with varying results. The theory is that they help the follicular epithelium mature and help correct the abnormal keratinization of the epidermis. Intralesional triamcinolone injections into the papules and plaques can also be an effective method of treating mild disease.
Severe disease is rarely responsive to medical therapy. Surgical options remain the best therapeutic choice. The goal is to remove the abnormal skin and close the wound under as little tension as possible. If the tension is too great, it is best to leave the wound open to granulate and heal by secondary intention. The scar that results is often better appearing than the thick, plaque-like scar that it is replacing.

Acute febrile neutrophilic dermatosis is an uncommon rash that most often is secondary to an underlying infection or malignancy. The diagnosis is made by fulfilling a constellation of criteria. Both clinical findings and pathology results are required to make the diagnosis in a patient with a consistent history.
Clinical Findings: Acute febrile neutrophilic dermatosis is often associated with a preceding infection. The infection can be located anywhere but most commonly is in the upper respiratory system. Females appear to be more likely to be afflicted, and there is no race predilection. Patients present with fever and the rapid onset of juicy papules and plaques. Because the papules can look as if they are fluid filled, they are given the descriptive term juicy papules . They can occur anywhere on the body and can be mistaken for a varicella infection. Patients also have neutrophilia and possibly arthritis and arthralgias. If this condition is associated with a preceding infection, it is usually self-limited and heals without scarring, unless the papules and plaques are excoriated or ulcerated by scratching. Variable amounts of pruritus and pain are associated with this skin disease. When one is evaluating a patient with this condition, a thorough history is required. A skin biopsy must be performed. A chest radiograph, throat culture, and urinalysis should be performed to assess for the possibility of bacterial infection.
Lymphoproliferative malignancies have also been seen in association with Sweet's syndrome. The malignancy often precedes the rash, and the skin disease is believed to be a reaction to the underlying malignancy. It is important to obtain specimens from these patients for histological evaluation and culture for aerobic, anaerobic, mycobacterial, and fungal organisms. The main differential diagnosis is between an infection and Sweet's syndrome in cases associated with a malignancy. The most common malignancy associated with acute febrile neutrophilic dermatosis is acute myelogenous leukemia. The prognosis in these cases is directly related to the underlying malignancy. Often, the skin disease continues to recur unless the malignancy is put into remission.
A few medications have also been shown to induce Sweet's syndrome, including granulocyte colony-stimulating factor (G-CSF), lithium, all- trans -retinoic acid, minocycline, and oral contraceptives.
Pathogenesis: The pathomechanism of Sweet's syndrome is theorized to involve the secretion of a neutrophilic chemoattractant factor, which causes massive amounts of neutrophils to migrate into the skin. The exact molecule responsible for the recruitment of neutrophils into the skin is unknown. Reports of exogenous use of G-CSF have led to the theory that it is responsible for the chemoattraction of neutrophils. Other chemoattractants are possible players in the pathogenesis, including interleukin-8.
Histology: Histological examination shows massive dermal edema with a dense infiltrate composed entirely of neutrophils. Varying amounts of leukocytoclasis are present. Subepidermal bulla formation is possible because of the extensive dermal edema. Special stains for microorganisms must be negative to exclude an infectious process, and these must be backed up with cultures to help disprove an infection, because the histological picture can mimic an infectious process.
Treatment: Treatment should be directed at the causative agent. Supportive care is needed for those with postinfectious Sweet's syndrome. Topical and oral steroids can dramatically shorten the course of the disease.
Sweet's syndrome that develops as a paraneoplastic process secondary to underlying leukemia should be treated with oral or intravenous steroids once an infectious process has been ruled out. This can result in a rapid response, but it is short lived once the steroids are removed. True remission occurs only if the cancer is treated and put into remission.

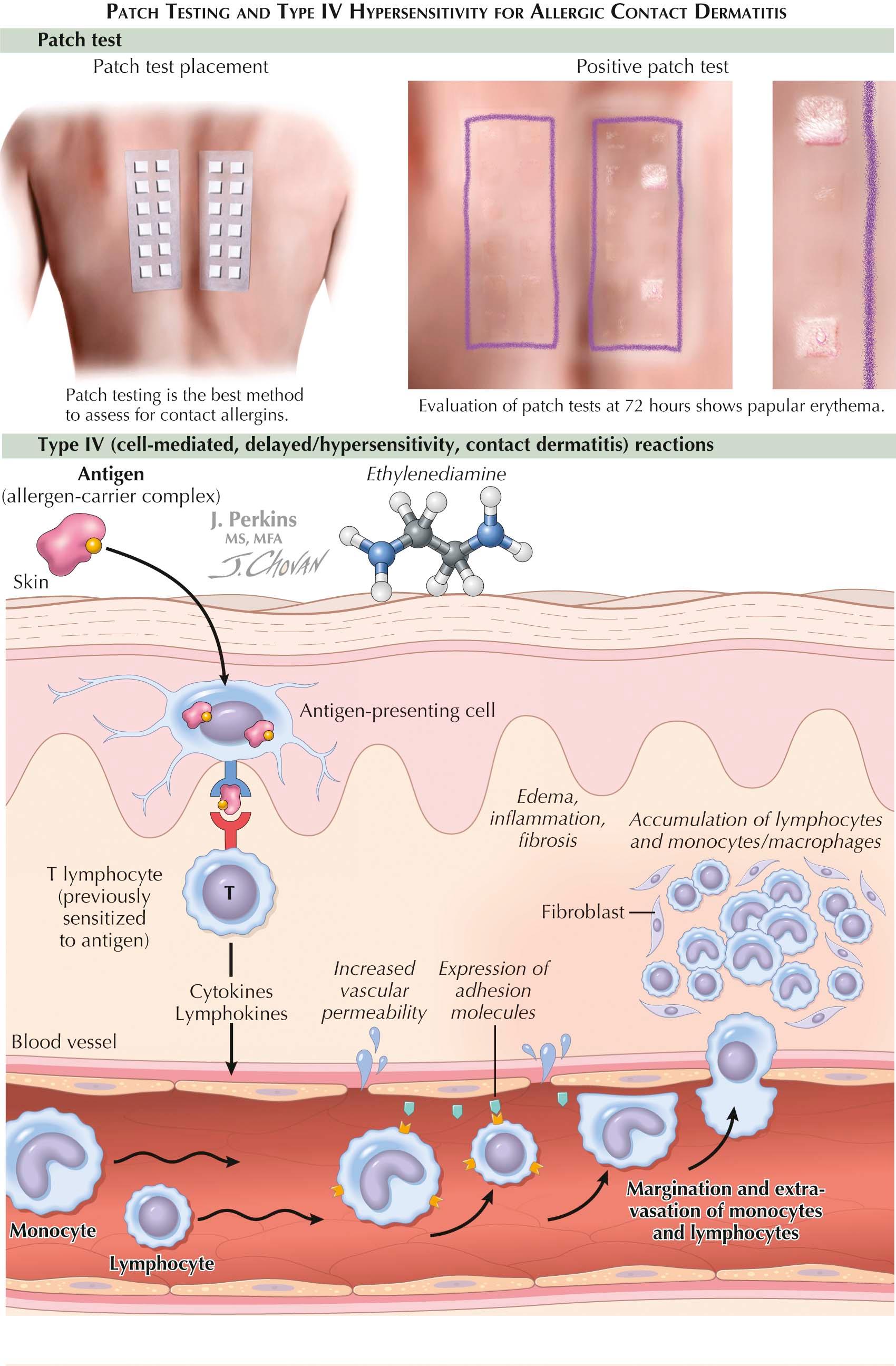
Allergic contact dermatitis is one of the rashes most frequently encountered in the clinician's office. It is responsible for a large proportion of occupationally induced skin disease. Urushiol from the sap of poison ivy, oak, or sumac plants is the most common cause of allergic contact dermatitis in the United States. The clinical morphology, the distribution of the rash, and results from skin patch testing are used to make the diagnosis. Patch testing is performed when the causative agent is unknown. Nickel has been the most frequent cause of positive patch testing in the world for years. Urushiol is not tested clinically, because almost 100% of the population reacts to this chemical.
Clinical Findings: Allergic contact dermatitis can manifest in a multitude of ways. The acute form may show linear streaks of juicy papules and vesicles. Variable amounts of surrounding edema can be seen. Edema is much more common in the loose skin around the eyelids and facial region. Chronic allergic contact dermatitis can manifest with red-pink patches and plaques with various amounts of lichenification. There are localized forms and generalized forms. One of the unique forms of allergic contact dermatitis is the scattered generalized form. Pruritus is an almost universal finding, and it can be so severe as to cause excoriations and small ulcerations.
The prototype of allergic contact dermatitis is the reaction to the poison ivy family of plants. After contact with this plant, urushiol resin is absorbed into the skin and initiates the immune system response to cause allergic contact dermatitis. The dose and the duration of contact with the allergen are important influences on the severity of the rash that develops. Between 3 and 14 days after exposure, the patient notices linear juicy papules and vesicles forming at the sites of contact. The most commonly affected areas are the extremities. Airborne contact dermatitis may be seen from burning of wood with the poison ivy vine present. These reactions are usually seen on skin that was not covered with clothing, and they can be very severe on the face and eyelids, often causing massive swelling and impeding vision.
The location of the dermatitis can be used as a clue to the diagnosis. A nurse with hand dermatitis may be allergic to a component of the gloves being worn occupationally. A young child with a lichenified rash around the umbilicus may be allergic to a metal component of a pant snap or zipper. The most common culprit in these cases is nickel. Finger dermatitis may be caused by the application of acrylic nails or nail polish. Allergic contact dermatitis can also be seen within the oral cavity, most commonly adjacent to dental amalgams or prostheses. Oral allergic contact dermatitis can mimic oral lichen planus. Lichen planus is usually widespread and affects the mucosa and gingiva both adjacent to and distant from any dental restorations.
The diagnosis in all these cases can be made based on patch testing. Chambers loaded with specific concentrations and amounts of known allergens are applied to the back of the individual. The patches are left on for 48 hours and then removed. After an hour, the first reading is made, based on the reaction seen under the chamber. Elevation of the skin or vesiculation is considered to be a positive reaction. The presence of only macular erythema needs to be interpreted cautiously but can be considered a positive result in certain situations. Pustular reactions are considered to be irritant reactions and not relevant. The patient must come back for a final reading 3 to 7 days after application of the patches. This is the most critical reading and gives the most valuable information.
Pathogenesis: Much is known about the mechanism of allergic contact dermatitis. This form of dermatitis requires a sensitization and elicitation phase for development. During the sensitization phase, the patient is exposed for the first time to the antigen. The antigen is absorbed through the skin and is phagocytosed by an antigen-presenting cell within the epidermis. The antigen-presenting cell internalizes the antigen and processes it within its lysosomal apparatus. The processed antigen is then sent to the cell surface and expressed on a human leukocyte antigen (HLA) molecule. The antigen-presenting cell migrates to the local draining lymph node and presents the antigen in association with the HLA molecule to T cells. The T cells recognize each individual antigen and proliferate locally, resulting in a clone of lymphocytes that recognize that specific antigen; these lymphocytes then remain ready for when the patient comes in contact with the same antigen in the future.
During the elicitation phase, the patient is reexposed to the antigen. The antigen-presenting cells again process the antigen and present it to the newly cloned lymphocytes, which migrate back to the skin and cause the clinical findings of edema, spongiosis, vesicles, and bullae. If the antigen is exposed in a chronic manner, the findings will be less acute in nature, and the typical findings of a chronic dermatitis are seen.
This entire process is dependent on the size and permeability of the antigen, the recognition and processing of the antigen by the antigen-presenting cell, and the complex interactions among multiple T and B cells. Antign-presenting cells and B cells are required for activation of the T cells and propagation of the allergic contact dermatitis.
Histology: The initial finding in acute allergic contact dermatitis is spongiosis of the epidermis with an associated superficial and deep lymphocytic infiltrate with scattered eosinophils. As the rash progresses, the spongiosis can worsen, and intraepidermal vesicles start to form. The vesicles may eventually coalesce into large bullae.
Chronic allergic dermatitis usually shows acanthosis with spongiosis and eosinophils within the infiltrate. A superficial and deep perivascular lymphocytic infiltrate is seen. Excoriations can also be appreciated.
Treatment: Acute localized allergic contact dermatitis can be treated with a potent topical steroid and strict avoidance of the offending agent. Oral sedating antihistamines work better for the pruritus than their nonsedating counterparts do. Soaks that help to dry the dermatitis are helpful and include aluminum acetate (Domeboro's solution). Because the most common culprit is the poison ivy plant, time should be taken to explain to the patient the appearance and nature of this plant. As a good rule of thumb, if a plant has three leaves, it could be poison ivy: “Leaves of three, let it be.” Allergic contact dermatitis that is widespread or that affects the eyelids, hands, or groin region can be treated with a tapering dose of oral corticosteroid over a 2- to 3-week period. If the steroid is tapered too quickly, the patient may experience a poststeroid flare of their dermatitis, which can be resistant to further corticosteroid therapy.
Patients who do not respond to these measures should undergo patch testing to determine whether another antigen is causing or provoking the dermatitis. Without the use of patch testing, the allergen will remain unknown and the dermatitis will persist. Not infrequently, patients are found to be allergic to a fragrance or preservative that is an ingredient in one of their personal care products. Once they stop using the product, the dermatitis finally resolves.


Atopic dermatitis is one of the most common dermatoses of childhood. It typically manifests in early life and can have varying degrees of expression. It is commonly associated with asthma and allergies. Most children eventually outgrow the condition. Atopic dermatitis has been estimated to affect up to 10% of all children and 1% of adults, and its prevalence has been steadily increasing. Patients frequently have a family history of atopic dermatitis, asthma, or skin sensitivity.
Clinical Findings: Atopic dermatitis typically begins early in life. There is no racial predilection. The clinical course is often chronic, with a waxing and waning nature. Infants a few months old may initially present with pruritic, red, eczematous patches on the cheeks and extremities as well as the trunk. The itching is typically severe and causes the child to excoriate the skin, which can lead to secondary skin infections. The skin of atopics is abnormally dry and is sensitive to heat and sweating. These children have difficulty sleeping because of the severe pruritus associated with the rash. During flares of the dermatitis, patients may develop weeping patches and plaques that are extremely pruritic and occasionally painful. With time, the patches begin to localize to flexural regions, particularly the antecubital and popliteal fossae. Severely afflicted children may have widespread disease. Patients with atopic dermatitis are more prone to react to contact and systemic allergens. Sensitivity to contact allergens is likely a consequence of the frequent use of topical medicaments and the broken skin barrier. This combination leads to increased exposure to foreign antigens that are capable of inducing allergic contact dermatitis. One should suspect a coexisting contact dermatitis if a patient who is doing well experiences a flare for no apparent reason or if a patient continues to get worse despite aggressive topical or oral therapy. Laboratory testing commonly shows an eosinophilia and an elevated immunoglobulin E (IgE) level.
Secondary infection is common in atopic dermatitis. It may manifest with the appearance of honey-colored, crusted patches in the excoriated regions, which indicates impetigo. It may also manifest as multiple follicle-based pustules, representing folliculitis, or as deep red, tender macules, indicating a deeper soft tissue infection. The rate of methicillin-resistant Staphylococcus aureus (MRSA) infection has increased in patients with atopic dermatitis at the same rate as in the general public. The rate of colonization of atopic patients is much higher than in normal controls, most likely because of the disruption of the underlying epidermis. Colonization in certain situations may lead to infection. Acquisition of a widespread herpesvirus infection can have severe and potentially life-threatening consequences. Atopics are much more prone than others to develop eczema herpeticum. The extensive areas of abnormal, broken skin provide the perfect environment for the development of this widespread viral infection.
Most childhood atopic dermatitis resolves spontaneously over time. It is estimated that 10% of cases will resolve by the age of 1 year, 50% by 5 years, 70% by 7 years, and so on. A small percentage of children with atopic dermatitis continue on with the rash into adulthood. These cases tend to be chronic in nature and to last for the patient's lifetime.
Pathogenesis: The cause of atopic dermatitis is unknown. Many exacerbating factors have been found. They include anything that irritates the skin, such as heat, sweating, stress, many chemicals, and various types of clothing. Atopic dermatitis is believed to be caused by an aberrant T-cell (Th2) response in the skin with elevated levels of Th2 cytokines. Interleukin-4 (IL-4), IL-5, and IL-13 are abnormally elevated. These cytokines are responsible for eosinophil production and recruitment and for IgE production. The concentrations of the Th1 cytokines (IL-12 and interferon-α) are below average in these patients. The reason for this response is unknown. Ultimately, the barrier of the epidermis is disrupted, and this is evident by the increase in transepidermal water loss, which can be measured.
Histology: A nonspecific lymphocytic infiltrate is seen, with associated exocytosis of lymphocytes into the epidermis with widespread spongiosis. Varying degrees of acanthosis and parakeratosis are seen. Often, bacterial elements are seen on the surface of the skin. Small intraepidermal vesicles may develop secondary to the massive spongiosis. Excoriations are frequently seen.
Treatment: Therapy consists of patient and family education about the natural history of the disease and the episodic waxing and waning. Bathing regimens must be thoroughly explained, and the use of soap should be discouraged. The patient should take shorter baths in lukewarm water, followed immediately by moisturization and application of topical steroid medications as appropriate. The intermittent use of moisturizers is also helpful. The use of topical immunomodulators, alternating with topical corticosteroids or alone, decreases the atrophogenic side effects of the topical corticosteroids. On occasion, oral steroids may be needed to calm the inflammation and give the patient some well-needed, albeit temporary, relief.
Most children do not need to avoid foods. If any question exists as to whether a food is potentially exacerbating the dermatitis, an allergist may be consulted to perform specific food allergy testing.
Prompt recognition of any bacterial or viral infection should lead to therapy that is not delayed. Impetigo, molluscum contagiosum, and eczema herpeticum are the three infections most commonly associated with atopic dermatitis. Of these, eczema herpeticum is the most important, and its recognition depends on a strong index of suspicion in any child with atopic dermatitis and new onset of a widespread, blistering rash. The differential diagnosis is varicella. A Tzanck test can help diagnosis the condition but cannot differentiate herpes simplex virus from varicella zoster virus. A viral culture or direct immunofluorescence antibody staining of blister fluid is required for differentiation.
Treatment is usually more successful in children than in adults. Occasionally in children and more commonly in adults, systemic therapies are used to keep the dermatitis under control. Oral antihistamines and immunosuppressive agents are not uncommonly required. A subset of patients respond to ultraviolet phototherapy, but most are not able to tolerate the warmth and sweating that is induced by the phototherapy unit. Oral immunosuppressants are used and include cyclosporine, azathioprine, and mycophenolate mofetil. These medications have severe potential side effects and should be administered only by experienced clinicians. Routine laboratory testing is required with all of these medications.

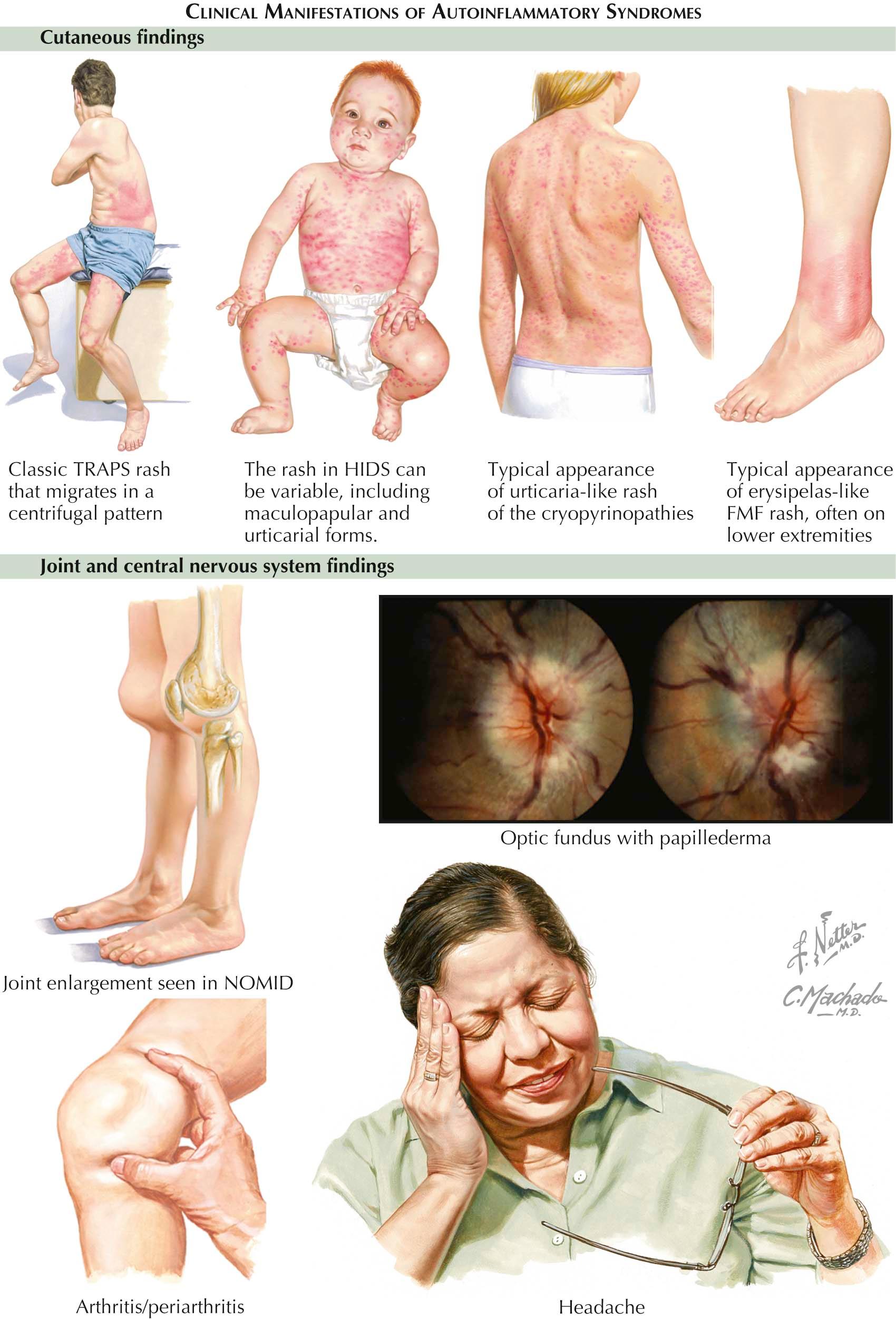
The autoinflammatory syndromes are a rare group of diseases for which the specific causes have been determined. The diseases in this category include hyper-immunoglobulin D (hyper-IgD) syndrome (HIDS), the cryopyrinopathies, familial Mediterranean fever (FMF), and tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS). The cryopyrinopathies are a group of conditions made up of Muckle-Wells syndrome, familial cold autoinflammatory syndrome (FCAS), neonatal-onset multisystem inflammatory disease (NOMID), and chronic infantile neurological cutaneous and articular syndrome (CINCA). These groupings were first proposed in the 1990s to bring together a collection of inflammatory disorders that are distinct in nature and pathophysiology from other forms of allergic, autoimmune, and immunodeficiency syndromes. Patients with these autoinflammatory diseases lack the autoreactive immune cells (T and B cells) as well as autoantibodies. The identification of specific genes that are defective and the roles played by those genes in the development of these disorders has been critical in increasing understanding of these diverse diseases. The common link in these conditions is the fact that they all represent abnormalities of the innate immune system.
Clinical Findings: HIDS is inherited in an autosomal recessive fashion. Patients present with fever, arthralgias, abdominal pain, cervical adenopathy, and aphthous ulcers. Skin findings are consistent with a cutaneous vasculitis with palpable purpura and purpuric macules and nodules. Patients develop attacks of these symptoms with some evidence of periodicity. The attacks can last from 3 to 7 days, and typically the first attack occurs within the first year of life. As the child ages, the frequency and the severity of the attacks lessen. No reliable trigger has been found that initiates the attacks, and patients are completely normal between attack episodes.
Within the group of cryopyrinopathies, the distinctions among Muckle-Wells syndrome, FCAS, NOMID, and CINCA are not clear, and many believe that they represent a phenotypic expression spectrum of the same condition. These very rare syndromes are all inherited in an autosomal dominant fashion. Patients present with recurrent fevers, arthralgias, myalgias, and varying degrees of ophthalmic involvement with conjunctivitis and anterior uveitis. The skin findings are typically generalized and consist of red, edematous papules and plaques. The rash can appear urticarial but is less pruritic. The attack episodes almost always last less than 24 hours. The trigger for FCAS is cold exposure, but the other conditions have no known precipitating factors. Twenty-five percent of patients with Muckle-Wells syndrome develop amyloidosis later in life, which may lead to chronic renal failure. The other conditions also have been reported to lead to amyloidosis, but much less commonly than Muckle-Wells syndrome. NOMID tends to be the most severe of the cryopyrinopathies. Patients with NOMID can develop aseptic meningitis and varying degrees of mental retardation along with hepatosplenomegaly. These patients can develop a characteristic overgrowth of cartilage around the knee that is quite noticeable on physical examination.
FMF is inherited in an autosomal dominant fashion. It is the most common of all the autoinflammatory syndromes. Patients experience attacks of fever and abdominal pain along with monoarthritis. Occasionally, pleuritis and pericarditis are also present. The skin findings consist of an erysipelas-like rash occurring almost exclusively on the lower extremities. Lesions of palpable purpura may also be present, indicating a cutaneous vasculitis. The attacks usually last less than 3 days, with a variable length of time between attacks. Some adults develop renal dysfunction due to amyloidosis.
TRAPS is inherited in an autosomal dominant pattern and also can occur sporadically. Patients develop attacks early in childhood, which consist of fever, abdominal pain, conjunctivitis, arthralgias, and migratory myalgias. The attacks last longer than in the other autoinflammatory syndromes. Each attack may last from days to weeks, with frequent recurrences. Attacks may be precipitated by varying amounts of stress, both physical and emotional. Again, the development of renal amyloidosis in adulthood has profound effects on the prognosis and is estimated to occur in 10% of TRAPS patients. Skin findings are characteristic and consist of migratory, pink to red patches and macules. Periorbital swelling may be prominent.
Histology: Each of the autoinflammatory skin lesions has a unique histology. The diagnosis cannot be made on the basis of histology alone, but histologic findings are used to rule out other conditions in the differential diagnosis and to help confirm the diagnosis of an autoinflammatory disease. Skin biopsies should be taken during acute attacks, when a rash is present.
Cutaneous biopsy specimens from patients with HIDS typically show a neutrophilic vasculitis. Neutrophils are found throughout the dermis. A skin biopsy from a patient with one of the cyropyrinopathies shows a neutrophilic perivascular infiltrate associated with diffuse dermal edema. NOMID and CINCA also exhibit a perivascular infiltrate of lymphocytes scattered within the neutrophilic infiltrate. FMF skin biopsies show a diffuse population of dermal neutrophils. TRAPS skin biopsies are nondescript and show a bland lymphocytic infiltrate in a dermal perivascular location. Biopsy of the periorbital edema shows a perivascular lymphocytic infiltrate and dermal edema.
Pathogenesis: Remarkable success has been achieved in deciphering the pathogenesis of these disease states, which are all interconnected through the innate immune system. The defective genes and the proteins that they encode have been determined. These proteins play a critical role in regulation of the innate immune system's inflammatory response. If they are defective, they cause varying amounts of dysregulation of neutrophils and other inflammatory cells. The innate immune system is nonspecific in nature and does not rely on antibody production. Various innate pattern recognition receptors (e.g., Toll-like receptors) are able to recognize foreign molecules and directly activate the innate immune system. The normal activation of the innate immune system allows for prompt recognition of foreign elements and a proper immune reaction to those elements. The autoinflammatory conditions have been discovered to involve defects in various components of the innate immune system.
HIDS is caused by a mutation in the MVK gene, located on chromosome 12, which encodes the protein mevalonate kinase. This gene helps regulate cholesterol synthesis, but it is also important for production of precursors that will ultimately be isoprenylated. The lack of these isoprenylated proteins leads to dysregulation of IL-1β and ultimately to the clinical findings of HIDS. All of the cryopyrinopathies are caused by a genetic defect of the NLRP3 gene located on chromosome 1. This gene, which is also called CIAS1 , encodes the protein cryopyrin. The defect allows for a gain in function of the cryopyrin protein, which results in hyperactivity of the inflammasome. The inflammasome is a cytoplasmic soluble conglomeration of various proteins that is part of the innate immune system and is constantly identifying foreign material. Its stimulation ultimately increases the activity of the caspase 1 protein and the production of IL-1β. FMF has been found to be caused by a defect in the MEFV gene, which encodes the pyrin protein. Pyrin is also a regulator of the inflammasome, and defects in pyrin result in increased levels of IL-1β. TRAPS is caused by a defective gene on chromosome 12 named TNFRSF1A . This gene encodes the 55-kd TNF receptor. The defect leads to excessive signaling due to serum TNF activation of the receptor.
Treatment: Therapy is specific to each syndrome. The molecular understanding of the pathogenesis has led to specific therapies. Because of their rarity, no randomized studies have been performed on the treatment of these conditions. HIDS has been successfully treated with nonsteroidal antiinflammatory drugs (NSAIDs), statin medications, and the interleukin antagonist, anakinra. The cryopyrinopathies have been treated with cold avoidance in the case of FCAS, and NSAIDs, oral steroids, anakinra, and other immunosuppressants have been tried. FMF has been treated with good success with colchicine, taking advantage of its antineutrophil effect. TRAPS has been successfully treated with etanercept or anakinra. Etanercept is believed to remove the soluble TNF that is responsible for activating the mutated receptor.


Human skin is exposed to the environment on a constant basis and encounters multiple threats, including arthropods of many varieties. Each species of arthropod can inflict its own type of damage to the skin; some bites are mild and barely noticeable, and others can be life-threatening. The most common bites are those of mosquitoes, fleas, bedbugs, mites, ticks, and spiders. Not only can these bites cause direct damage to the skin, but these organisms may have the ability to transmit infectious diseases such as Lyme disease, leishmaniasis, and rickettsial diseases.
Clinical Findings: Mosquitoes are prominent insects in the spring, summer, and early fall seasons. In warmer climates, they can be seen year round. Their bite is often not noticed until after the mosquito has gone. The recently bitten person is left with a pruritic urticarial papule that typically resolves by itself within an hour or so. Some individuals are prone to severe bite reactions and develop warm, red papules and nodules that can last for a week or two and can be associated with regional lymphadenopathy. Mosquitoes are essentially a nuisance for the most part, but in some areas of the world they are the major vectors for transmission of malaria and encephalitis viruses. Sand flies are similar, but they are the major vector for leishmaniasis.
Fleas have been around since before the beginning of human civilization and were responsible in the Middle Ages for helping transmit the bubonic plague, which killed millions of people. Fleas are most commonly seen in households with pets. Individuals can be bitten after the pet transfers the fleas to bedding, carpeting, or clothing. Characteristic bites occur in groups of three, referred to as “breakfast, lunch, and dinner.” Flea eggs can lay dormant for years, only to reactivate in response to movement and vibration that indicate a meal is likely to be nearby. Many flea bites occur around the ankles of adults; the fleas jump from the carpeting to the ankles, take their meal, and leave. The typical skin lesion is a small papule with a central punctum. It is self-resolving. Fleas have been known to carry organisms responsible for infectious diseases, including Yersinia pestis (bubonic plague) and Rickettsia typhi (murine typhus).
Bedbugs (Cimex lectularius) have made a resurgence in the United States. The are ubiquitous insects that can live in any area of the country. Households, hotels, and other sleeping quarters become infested with colonies of bedbugs. They emerge in the night, typically 1 to 2 hours before dawn, and search for a blood meal. They find their victim asleep and feed for a few minutes before retreating back to the nest. The nest is almost never in the bed; it is most likely to be located within the baseboard molding or floor boards. In the morning, the afflicted individual awakens with one to hundreds of bites. Most are small papules with a central punctum. Depending on the species of bedbug, a more inflammatory response may occur, causing vesiculation and bullae. Bedbugs have been reported to transmit hepatitis B virus.
Encounters with the large mite family of organisms are more likely to occur in the summer months in northern latitudes but can occur at any time of the year in the southern regions. The term chigger refers to the larval phase of the Trombiculidae family of mites; it is one of the most common and well-recognized causes of human bites. Chiggers are small red mites, so small that they are not felt, and they bite quickly. They usually leave pinpoint red papules that can be numerous and can cause severe pruritus. Many other mites are present in the environment and can cause similar reactions.
Most ticks bite and feed for up to 24 hours before falling off after receiving their blood meal. They can leave a tick bite granuloma, which is a small red papule with a central punctum, at the site of the bite. Many methods have been used to remove ticks; most can result in more skin damage than the actual tick bite. These methods include burning the end of the tick with a cigarette or a match, an approach that is more likely to cause a skin burn than it is to remove the tick. The best method of removal is to grab the tick as close to the surface of the skin as possible and gently pull in a direction perpendicular to the skin. If the mouthparts are left embedded in the skin, a small punch biopsy can be performed to remove the remaining parts. Ticks are well known to transmit many infectious diseases, including Lyme disease and Rocky Mountain spotted fever.
Most spider bites are caused by jumping spiders. As with all spiders, bites frequently occurs after the spider's web or nesting location is disturbed. The bites can be painful and can leave erythema and a papule or nodular reaction. On occasion, these bites develop secondary cellulitis. Two spiders are unique in their potential to cause severe human disease: the black widow spider (Latrodectus mactans) and the brown recluse spider (Loxosceles reclusa) .
The black widow spider is a web-weaving spider that paralyzes its prey with a potent neurotoxin called latrotoxin. The venom causes massive release of acetylcholine from nerve endings. In humans, this can lead to pain, fever, and symptoms of an acute abdomen.
The brown recluse spider is a solitary stalking spider that lives in dark, hidden locations. It is not aggressive and typically bites only when a human accidentally disturbs its location. The toxin released in its venom contains a mixture of sphingomyelinase-D, hyaluronidases, proteases, and esterases. Sphingomyelinase-D is the major component that is believed to be responsible for most of the tissue damage caused by the spider's bite. It can cause severe pain and aggregation of platelets and red blood cells, resulting in intravascular clotting with resultant necrosis of the skin. The characteristic pattern seen on the skin is a central bluish region with necrosis and coagulation, a surrounding vasoconstricted area that appears to be blanched white and a peripheral rim of erythema. This has been termed the “red, white, and blue” sign of a brown recluse bite. Some bites can progress rapidly and cause severe necrosis of the skin requiring surgical debridement.
Histology: Most bite reactions are not biopsied, because they are typically diagnosed clinically. The histological findings for most bug bites are very similar. There is a superficial and deep inflammatory infiltrate with many eosinophils. Superficial necrosis of the epidermis may be seen at the site of the bite. Occasionally, tick mouth parts are located in the biopsy specimen. Brown recluse spider bites show intravascular thrombosis and necrosis of the skin.
Treatment: The treatment of most bites is supportive. Pruritus can be treated with a potent topical corticosteroid and an oral antihistamine. Avoidance is the most important preventive measure. Areas of standing water provide breeding grounds for mosquitoes and should be drained routinely. Pets should be groomed and treated with preventive tick and flea medications. Flea and bedbug infestations should be treated by a professional exterminator. Proper use of bug sprays containing DEET ( N,N -diethyl- m -toluamide) and staying in the center of wooded trails can help decrease one's chance of being bitten. In endemic areas, any patient with a deer tick bite that has lasted longer than 24 hours should be considered for prophylactic therapy for Lyme disease.
Narcotics (for pain control) and antivenin have been used to treat black widow spider bites and have been helpful. The antivenin is derived from horse serum, and there is a risk of an allergic reaction in susceptible patients. Brown recluse spider bites have been treated with many agents, including dapsone, to try to mitigate some of the inflammation-induced skin damage. Recognition of these spiders and avoidance is critical.

Calciphylaxis (calcific uremic arteriolopathy) results from deposition of calcium in the tunica media portion of the small vessel walls in association with proliferation of the intimal layer of endothelial cells. It is almost always associated with end-stage renal disease, especially in patients undergoing chronic dialysis (either peritoneal dialysis or hemodialysis). It has been reported to occur in up to 5% of patients who have been on dialysis for longer than 1 year. Calciphylaxis typically manifests as nonhealing skin ulcers located in adipose-rich areas of the trunk and thighs, but the lesions can occur anywhere. They are believed to be caused by an abnormal ratio of calcium and phosphorus, which leads to the abnormal deposition within the tunica media of small blood vessels. This eventually results in thrombosis and ulceration of the overlying skin. Calciphylaxis has a poor prognosis, and there are few well-studied therapies.
Clinical Findings: Calciphylaxis is almost exclusively seen in patients with chronic end-stage renal disease. Most patients have been on one form of dialysis for at least 1 year by the time of presentation. The initial presenting sign is that of a tender, dusky red to purple macule that quickly ulcerates. The ulcerations have a ragged border and a thick black necrotic eschar. The ulcers tend to increase in size, and new areas appear before older ulcers have any opportunity to heal. Ulcerations begin proximally and tend to follow the path of the underlying affected blood vessel. Their most prominent location is within the adipose-rich areas of the trunk and thighs, especially the abdomen and mammary regions. Patients often report that ulcerations form in areas of trauma. The main differential diagnosis is between an infectious cause and calciphylaxis. Skin biopsies and cultures can be performed to differentiate the two. Skin biopsies are diagnostic. Radiographs of the region often show calcification of the small vessels, and this can be used to support the diagnosis. Patients who develop calciphylaxis have a poor prognosis, with the mortality rate reaching 80% in some series. For some unknown reason, those with truncal disease tend to survive longer than those with distal extremity disease. Complications caused by the chronic severe ulcerations (e.g., infection, sepsis) are the main cause of mortality.
Laboratory findings often show an elevated calcium × phosphorus product. A calcium × phosphorus product greater than 70 mg 2 /dL 2 appears to be an independent risk factor for development of calciphylaxis. Other risk factors are obesity, hyperparathyroidism, diabetes, and the use of warfarin. Elevated parathyroid hormone (PTH) levels are often found in association with calciphylaxis. The exact role that PTH plays is unknown, but it has been reported that parathyroidectomy, a standard treatment for calciphylaxis in the past, is not an effective means of therapy. PTH may play a role in starting the disease, but it does not appear to be necessary to exacerbate or cause continuation of calciphylaxis.
Pathogenesis: The exact mechanism of calcification of the tunica media of blood vessels in calciphylaxis is not completely understood. The fact that it is seen almost exclusively in patients undergoing chronic dialysis therapy has led to many theories on its origin. The final mechanism is a hardening of the vessel wall with calcification and intimal endothelial proliferation that leads to rapid and successive thrombosis and necrosis.
Histology: The main finding is of calcification of the medial section of the small blood vessels in and around the area of involvement. Thrombi within the vessel lumen are often observed. Intimal layer endothelial proliferation is prominent. The abnormal calcification can easily be seen on hematoxylin and eosin staining.
Treatment: No good therapy exists for calciphylaxis. Aggressive supportive care and early treatment of superinfections are critical. Surgical debridement of wounds is necessary to remove necrotic tissue that provides a portal for infection. Renal transplantation offers some hope for cure. Treatment with sodium thiosulfate has shown success in some anecdotal reports, but this is not a universal cure. The newer bisphosphonate medications have also been used with limited success. Parathyroidectomy may help initially with the ulcerations, but it does not decrease mortality.



Lupus erythematosus is a multisystem, idiopathic connective tissue disease that can have variable and unique clinical cutaneous findings. Cutaneous lupus may be considered as a spectrum of skin disease. Many variants have been described. Discoid lupus, subacute cutaneous lupus, tumid lupus, lupus panniculitis, neonatal lupus, lupus chilblains, and systemic lupus erythematosus (SLE) all have morphologically distinctive cutaneous findings. Lupus is a heterogeneous disease with a wide continuum of clinical involvement, from purely cutaneous disease to life-threatening SLE. The cutaneous findings are often the first presenting signs, and recognition of the skin manifestations can help make the diagnosis of lupus.
SLE is the most severe form of lupus. Its clinical course and outcome vary, from mild forms to severe, life-threatening variants. In the most severe cases, the pulmonary, cardiac, neurological, and connective tissue and integumentary systems are affected. Death may occur from renal failure. Severe arthritis and skin findings are often present. SLE is diagnosed by fulfillment of criteria that have been established by the American College of Rheumatology. Variations in meeting these criteria from one patient with SLE to the next are responsible for the varying clinical spectrum of disease.
Patients with lupus can have many laboratory abnormalities. These include anemia of chronic disease and an elevated erythrocyte sedimentation rate. Antinuclear antibodies (ANA) are found in some subsets of lupus, with almost 100% of patients with the systemic form testing positive for ANA. Many other, more specific antibodies are found in patients with SLE, including anti-Smith antibodies and anti–double-stranded DNA antibodies. Patients with renal disease often have hypertension, elevated protein levels in their urine, and an elevated creatinine level.
Clinical Findings: Many variants of cutaneous lupus exist, each with its own morphological findings. Lupus is more common in women; it can be seen at any age but is most frequently observed in early adulthood. However, lupus is common enough that it is not infrequently seen in males. Neonatal lupus is a rare form that occurs in neonates born to mothers with lupus.
Discoid lupus is one of the easiest forms of cutaneous lupus to recognize. It is most commonly found on the head and neck region and has a tendency to be present within the conchal bowl of the ear. Lesions are often found in patients with SLE. Discoid lupus may occur as an entirely separate disease with no other systemic or clinical findings of lupus. Fewer than 10% of these patients eventually progress to the systemic form of lupus. Discoid lesions are exacerbated by sun exposure, more specifically by exposure to ultraviolet A (UVA) light. The lesions tend to have an annular configuration with varying amounts of scale. The lesions can produce alopecia, and there is almost always some amount of atrophy present. Follicular plugging is commonly seen in discoid lupus. It is noticed clinically as a dilation of the follicular orifices. Follicle plugs can also be seen by gently removing the scale from a discoid lesion. On close inspection of the inferior side of the scale, one will notice minute keratotic follicular plugs. This finding is specific for discoid lupus and has been termed the “carpet tack sign,” because it resembles tiny outreaching tacks. This sign can be easily missed if the scale is removed too quickly or not inspected closely enough. Discoid lesions in darker-skinned individuals may also have varying amounts of hyperpigmentation. Most patients have some erythema and hyperpigmentation. Most patients present with a few discoid lesions and are said to have localized discoid lupus. Those rare patients with widespread disease have generalized discoid lupus. This variant is rare, and such patients are much more likely than those with localized disease to go on to fulfill the criteria for SLE at some point. The alopecia seen in discoid lupus is scarring in nature, and the hair that has been lost will not regrow even with aggressive therapy. Alopecia can be life-altering and can cause significant psychological morbidity.
Subacute cutaneous lupus erythematosus is seen in a subset of patients and has a higher incidence of developing into full-blown SLE compared with other forms of cutaneous lupus. There are variants of subacute cutaneous lupus, with the annular form and the papulosquamous form being the two most common and most important to recognize. The annular form manifests with pink to red annular patches that slowly expand and coalesce into larger, interconnected polycyclic patches. They occur most commonly on sun-exposed skin of the face and upper trunk. The papulosquamous version also manifests in sun-exposed regions. It appears as smaller, pink-red patches with overlying scale. Both forms are exacerbated by sun exposure and are pruritic. They heal with no scarring.
Neonatal lupus is an uncommon form of lupus that can manifest with or without cutaneous findings. However, cutaneous findings are the most universal clinical finding in neonatal lupus, occurring in more than 90% of those affected. The most common scenario is a child born to a mother who has not yet been diagnosed with lupus. Neonatal lupus can manifest with varying degrees of congenital heart block, and this is the most serious sequela. Some children require a pacemaker to control the arrhythmia. Thrombocytopenia is also one of the more frequent effects of neonatal lupus. Neonatal lupus is directly caused by the transplacental migration of anti-Ro (anti-SSA) antibodies and, to a lesser extent, anti-La (anti-SSB) antibodies. The antibodies are only transiently present, because the newborn does not produce any new antibodies. Therefore, neonatal lupus improves over time, and most children have no long-term difficulties. The cutaneous findings in neonatal lupus include pink to red patches or plaques, predominantly in a periorbital location. The rash resolves with time, and if any residual skin finding remains, it is that of fine telangiectases in the location of the patches and some fine atrophy. The telangiectases and atrophy tend to improve as the child enters adulthood.
Lupus panniculitis (lupus profundus) is a rare cutaneous manifestation of lupus. It manifests as a tender dermal nodule, more commonly in women. A large percentage of patients with lupus panniculitis have been reported to go on to develop SLE. The overlying skin may appear slightly erythematous to hyperpigmented, but there is no appreciable surface change. The dermal nodules tend to slowly enlarge with time. The diagnosis can be made only by biopsy, because the clinical picture is not specific. Biopsies of these dermal nodules are best performed with an excisional technique to obtain sufficient tissue for diagnosis. The inflammation is entirely confined to the subcutaneous tissue. The histological differential diagnosis of lupus panniculitis is often between lupus and cutaneous T-cell panniculitis. The diagnosis requires the use of both clinical and histological information. The histological evaluation often requires immunohistochemical staining to help differentiate the lesions from those of other mimickers. Lesions of lupus panniculitis often heal with atrophic scarring.
Tumid lupus is a rare clinical variant of cutaneous lupus that typically manifests as a red dermal plaque on a sun-exposed surface of the skin. Clinically, it can appear similar to polymorphous light eruption, lymphoma, pseudolymphoma, or Jessner's lymphocytic infiltrate. The plaques are exacerbated by ultraviolet light exposure. They are frequently asymptomatic to slightly tender but rarely pruritic. They tend to wax and wane, with the worst outbreaks occurring in the springtime and remissions in the winter. Histologically, the infiltrate has been found to be more of a CD4+ T-cell infiltrate.
Lupus chilblains is a unique form of Raynaud's phenomenon, and it is identical in clinical presentation to pernio. It may be that this is just pernio occurring in a patient with lupus. Lupus chilblains and pernio manifest typically on the distal extremities, the toes being the most commonly affected region. The patient develops tender, cold, purplish papules and plaques. The rash is exacerbated by cold and wet environments. Treatment includes keeping the regions dry and warm by avoiding cold exposure. Patients diagnosed with pernio probably should undergo screening for lupus, because a small percentage of them actually have lupus chilblains. Histological evaluation of lupus chilblains shows a dense lymphocytic infiltrate with some areas of thrombosis of small vessels and a lymphocytic vasculitis.
The cutaneous findings seen in SLE are vast and can overlap with other forms of cutaneous lupus. Although the systemic findings are responsible for the morbidity and mortality, the cutaneous findings are often the presenting sign, and, if the clinician is aware, they can help make the diagnosis. The most important of the cutaneous skin findings in SLE is the malar rash. This rash manifests as a tender, pink-to-red plaque or patch on the cheeks and nose, mimicking the shape of a butterfly; hence, it has been termed the “butterfly rash of lupus.” It is commonly mistaken for rosacea, and vice versa. Rosacea typically affects a wider area of skin and is associated with more telangiectases and papulopustular lesions. The malar rash of lupus also spares the nasolabial fold, which is an important clinical finding and a discriminating objective discovery. It is typically more prominent during systemic flares of the underlying SLE, and patients can appear very ill. Patients are exquisitely photosensitive, and the rash is exacerbated by exposure to ultraviolet light.
Discoid lupus is also seen as a manifestation of systemic lupus, and it has the same clinical appearance as described earlier. Raynaud's phenomenon is well described, and a high percentage of patients with SLE report those symptoms. Alopecia was long used to help make the diagnosis of lupus. It is no longer part of the diagnostic criteria, but it can have significant psychological impact on the patient. Nail and capillary nail fold changes are seen if looked for. The true incidence of these findings is unknown. Nail fold telangiectases and erythema are the two most common nail findings. Nail pitting, ridging, and alterations in the color of the lunula have also been reported. Lupus patients with nail changes have been found to have a higher incidence of mucosal ulcerations, which are another of the mucocutaneous findings of SLE. Livedo reticularis is a fishnet-like pattern found typically on the lower extremities; it is a nonspecific finding but has been reported commonly in lupus. It also occurs in many other skin and systemic diseases.
Histology: The histological findings in all forms of lupus are similar, with specific forms having some unique findings. Most forms show an interface dermatitis with hydropic changes in the basilar layer of the epidermis. A superficial and deep periadnexal lymphocytic infiltrate is almost universally seen. Other connective tissue diseases (e.g., dermatomyositis) can have similar histological findings. Discoid lupus may show scarring, atrophy, and follicular plugging along with these other findings. Lupus panniculitis is unique in that the inflammation is localized to the subcutaneous tissue. The diagnosis of lupus panniculitis is difficult and requires a host of special stains and clinical pathological correlation.
Treatment: The treatment of cutaneous lupus is difficult and must be tailored to the patient and the specific form of lupus. Potent topical corticosteroids may work for a tiny lesion of discoid lupus, but they are not effective in lupus panniculitis. Universal treatment of cutaneous lupus requires sun protection and sunscreen use. The sunscreen used should block in the UVA range, because this is the most active form of ultraviolet light that exacerbates lupus. Smoking should be ceased immediately, and patients should be screened routinely by their family physician or rheumatologist for progression of the disease.
Specific therapies for cutaneous lupus include oral prednisone and hydroxychloroquine or chloroquine as the typical first-line agents. If these are unsuccessful, quinacrine can be added. Other agents that have been reported to be effective include dapsone, isotretinoin, and methotrexate.

Cutis laxa is an unusual skin disease with multisystem complications. It has highly characteristic cutaneous findings. Laxity of the skin is the hallmark of this disease. The skin becomes easily stretched, and there is little elastic rebound. As patients age, gravity alone can make the skin droop to a disfiguring degree. Some forms of cutis laxa are incompatible with life, and those affected die in infancy. Many variants of cutis laxa have been described. With the discovery of the responsible gene defects, the phenotypes of this disease that are seen clinically have been better defined on the genetic level. Acquired variants of cutis laxa have been described.
Clinical Findings: Cutis laxa has no sexual or racial predilection. The cutaneous hallmark of the disease is loose, hanging skin with a lack of elasticity. The skin can be pulled with little resistance; the normal return of the skin to its preexisting state is delayed. The skin in the axillae and groin folds is prominently affected, as is the facial skin. The face is said to take on a “hound dog” appearance. All skin is involved to varying degrees, but the effects are most noticeable in areas of the face and in the skin folds. The overlying epidermis is completely normal, and the adnexal structures are spared.
Internal manifestations are variable and are more common with the autosomal recessive forms of the disease. The pulmonary, cardiovascular, and gastrointestinal systems can be affected by fragmentation or loss of elastic tissue, leading, respectively, to emphysema, aneurysms, and diverticula.
Those with the autosomal dominant form appear to have normal life spans, whereas those with the other variants have significantly shortened life spans secondary to severe systemic involvement.
Pathogenesis: Many modes of inheritance have been reported for cutis laxa, including autosomal recessive, autosomal dominant, and X-linked recessive forms. The X-linked form is now considered to be the same disease as Ehlers-Danlos syndrome IX. This form is caused by a defect in a copper-dependent adenosine triphosphatase (ATPase) protein found within the Golgi apparatus.
There are two autosomal recessive variants of cutis laxa. The autosomal recessive variant type I is extremely rare, and those afflicted typically die early in infancy from severe pulmonary and multisystem failure. Autosomal recessive type I cutis laxa has been found to be caused by a defect in the fibulin-5 gene (FBLN5) . The product of this gene is critical in producing functional elastic fibers. Its absence is incompatible with life. Type II autosomal recessive cutis laxa is more commonly encountered than type I. The genetic defect in type II cutis laxa has yet to be defined. Patients with type II experience developmental delay and have varying amounts of joint laxity.
The most frequently seen form of cutis laxa is the autosomal dominant form, which is caused by a defect in the elastin gene (ELN) . Many different mutations in this gene have been described, and they lead to slightly different phenotypes of the disease.
All of these gene defects lead to abnormalities in the elastic fiber protein, resulting in elastolysis. Various defects lead to different irregularities in the elastic fibers, but the end result in all forms is seen clinically as cutis laxa.
Histology: Histological examination of skin biopsies from patients with cutis laxa reveals varying degrees of elastic fiber damage and/or loss. The best way to appreciate this is with special staining to highlight elastic tissue. In some cases, there is a complete loss of elastic fibers; in others, fragmented and reduced amounts of elastic tissue are seen.
Treatment: The main goals of therapy is to screen for underlying cardiac or gastrointestinal abnormalities and for the possibility of aortic aneurysm or gastrointestinal diverticula formation. There is no medication that can reverse the genetic defect, and no gene replacement therapy is available. Excessive skin can be surgically removed to improve functionality and cosmesis.

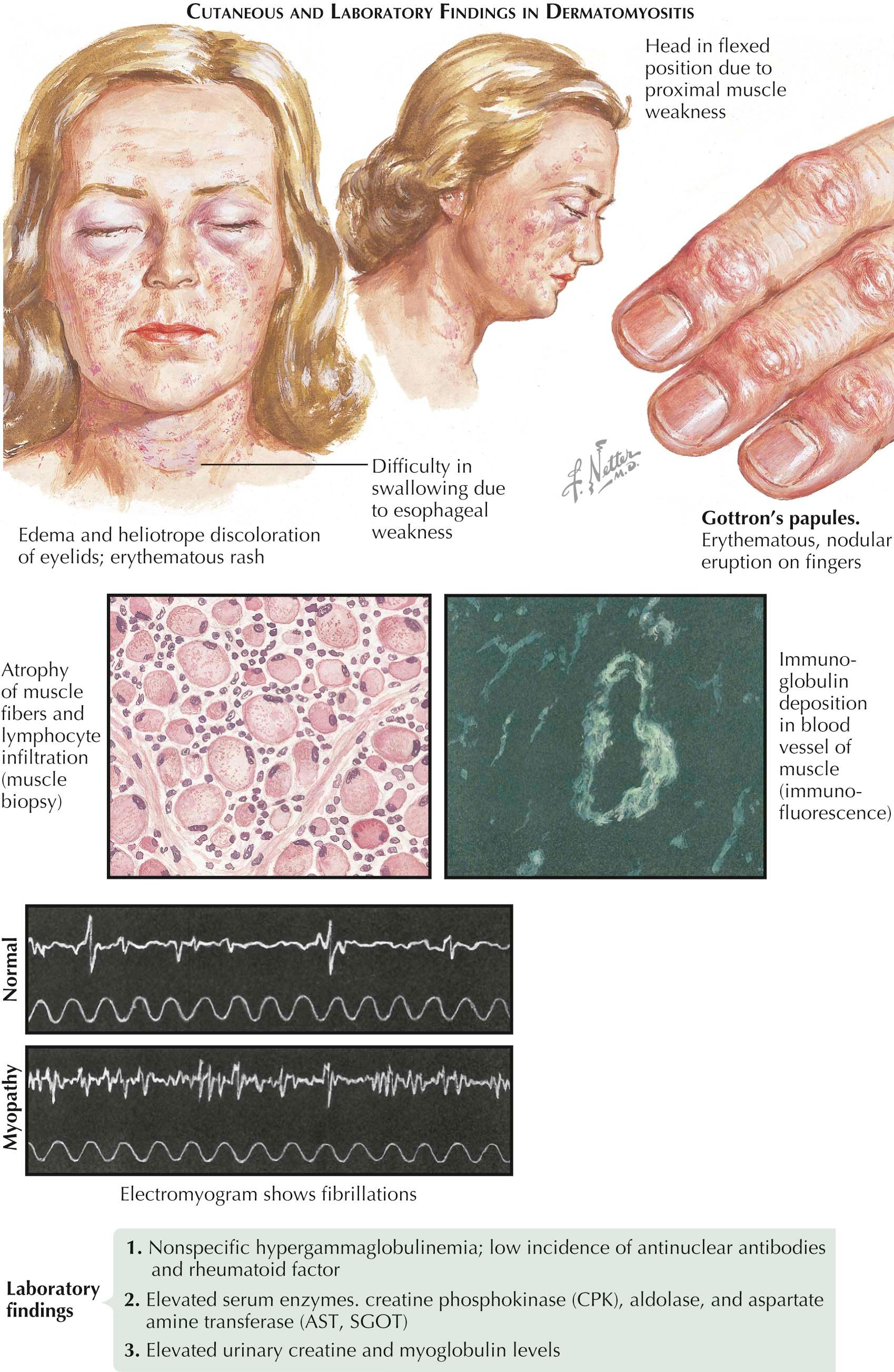
Dermatomyositis is a chronic connective tissue disease that can be associated with an underlying internal malignancy. This connective tissue disease shares similarities with polymyositis, but the latter has no cutaneous findings. Up to one third of patients with dermatomyositis have an underlying malignancy. The myositis is often prominent and manifests as tenderness and weakness of the proximal muscle groups. The pelvic and shoulder girdle muscles are the ones most commonly affected. Dermatomyositis sine myositis is a well-recognized variant that has only the cutaneous findings; evidence of muscle involvement is absent.
Clinical Findings: Dermatomyositis has a bimodal age of onset, with the most common form occurring in the female adult population, usually between the ages of 45 and 60 years, and a smaller peak in childhood at about 10 to 15 years of age. African Americans are affected three to four times more often than Caucasians. Dermatomyositis has an insidious onset, with the development of proximal muscle weakness in association with various dermatological findings. Skin findings start slowly and are nonspecific at first. Usually, there is some mild erythema on the hands and sun-exposed regions of the head and neck. Over time, the more typical cutaneous findings become evident. Pruritus is a common complaint, and patients not infrequently complain of severe scalp pruritus well before any signs or symptoms of dermatomyositis appear.
The heliotrope rash of dermatomyositis is one of the most easily recognized and specific findings. It is manifested by periorbital edema and a light purple discoloration of the periorbital skin. The skin is tender to the touch. Hyperemia of the nail beds and dilated capillary loops are noticeable and are similar to those seen in progressive systemic sclerosis or lupus erythematous. The dilated capillary loops are best appreciated with the use of a handheld dermatoscope that serves to magnify the region of interest.
Purplish to red, scaly papules develop on the dorsum of the hands overlying the joints of the phalanges. These are not Heberden's nodes, which are a manifestation of osteoarthritis seen as dermal swellings overlying the distal interphalangeal joints. The papules seen in dermatomyositis have been termed Gottron's papules . Gottron's papules may be seen overlying any joint on the hands, as well as other joints such as the elbows and knees. The skin findings on the dorsal hands have led to the term “mechanic's hands.” This refers to the ragged appearance of the hands in dermatomyositis; they resemble the hands of a mechanic that have suffered chronic trauma, abrasions, and erosions secondary to the occupation.
The “shawl sign” is a cutaneous finding seen on the upper back and chest. The shawl sign is so named because the location is in the same area that would be covered by a shawl garment. The skin has poikilodermatous macules and patches. There is a varying amount of skin atrophy with telangiectases, mottled hyperpigmentation and hypopigmentation, and erythema of the involved region.
Patients with dermatomyositis also complain of photosensitivity and notice a flare of their skin disease with ultraviolet light exposure. Children with dermatomyositis are much more prone to develop calcinosis cutis than their adult counterparts, and approximately 50% of all children with dermatomyositis will develop this feature. Calcinosis cutis manifests as tender dermal nodules or as calcifications along the muscle fascia. Leukocytoclastic vasculitis also is seen much more frequently in juvenile dermatomyositis than in the adult form.
Dermatomyositis is a multisystem disorder. Diagnostic criteria have been established by the American College of Rheumatology. They are based on the presence of clinical, laboratory, and histological findings. Not all patients have all aspects of the disease, and the diagnosis is based on the number of the criteria fulfilled.
Inflammation of the proximal muscle groups has been well described. Patients often complain of difficulty in standing from a sitting position or in raising their hands above their heads. Patients have elevated serum concentrations of creatinine kinase, aldolase, and lactate dehydrogenase. This is indicative of muscle inflammation and breakdown. An electromyogram (EMG) can be used to evaluate the weakness and to differentiate a nerve origin from a muscle origin. A muscle biopsy, most commonly of the deltoid muscle, shows active inflammation on histological examination.
This disease can rarely manifest with severe, diffuse interstitial pulmonary fibrosis. Patients with pulmonary fibrosis most often test positive for the anti-Jo1 antibody. Anti-Jo1 antibodies have been found to be targeted against the histidyl–transfer RNA synthetase protein. Overall, it is an uncommon finding except in dermatomyositis patients with pulmonary disease. More than 75% of patients with dermatomyositis test positive for antinuclear antibodies (ANA). Those with malignancy-associated dermatomyositis typically do not develop pulmonary fibrosis, and those with pulmonary fibrosis do not develop a malignancy.
The malignancy most commonly associated with dermatomyositis is ovarian cancer. Many other malignancies have been seen in association with dermatomyositis, including breast, lung, lymphoma, and gastric cancers. Malignancy is seen before the onset of the rash in about one third of the cases, concurrently with the rash in one third, and within 2 years after diagnosis of the dermatomyositis in one third. After the diagnosis of dermatomyositis, it is imperative to search for an underlying malignancy and to perform age-appropriate cancer screening. Childhood dermatomyositis is rarely associated with an underlying malignancy.
Pathogenesis: The exact etiology of dermatomyositis is unknown. It has been theorized to occur secondary to abnormalities in the humoral immune system. The precise mechanism is under intense research.
Histology: Histological examination of a skin biopsy specimen shows an interface lymphocytic dermatitis. Hydropic change is seen scattered along the basilar cell layer. The epidermis has varying degrees of atrophy. A superficial and deep periadnexal lymphocytic infiltrate is common. The presence of dermal mucin in abundance is another histological clue to the diagnosis. A muscle biopsy often shows atrophy of the involved muscle with a dense lymphocytic infiltrate.
Treatment: There is no known cure for dermatomyositis, although some cases spontaneously remit. Cases associated with an underlying malignancy have been shown to go into full remission with cure of the underlying cancer. Relapse of dermatomyositis in these patients should prompt the clinician to search for a recurrence of their malignancy. Initial treatment is usually with prednisone, which acts as a nonspecific immunosuppressant. The addition of a steroid-sparing agent is almost always needed to avoid the long-term side effects of prednisone. Some patients require a smaller dose of prednisone along with a steroid-sparing agent to keep the disease at bay. Many steroid-sparing agents have been used, including hydroxychloroquine, quinacrine, cyclosporine, intravenous immunoglobulin (IVIG), azathioprine, and methotrexate, all with variable success. Combination therapy is the norm.
The use of sun protection and sunscreen cannot be overemphasized. Topical corticosteroids help relieve the itching and decrease some of the redness. The treatment of juvenile dermatomyositis is similar. It is believed to have a better prognosis, because few cases are associated with an underlying cancer. It is thought that early treatment of juvenile dermatomyositis decreases the risk of developing severe calcinosis cutis during the course of the disease.

Disseminated intravascular coagulation (DIC) is a serious, life-threatening condition of the blood clotting system that can be caused by myriad insults to the body. It has a grave prognosis unless caught and treated early in the course of disease. Skin manifestations occur early and continue to progress unless the patient recovers. The skin lesions may lead to gangrene and secondary infection, further worsening the prognosis. DIC is seen as an end-stage process, caused by the consumption of blood clotting factors, that results in uncontrolled clotting and bleeding occurring simultaneously.
Clinical Findings: DIC occurs in males and females with equal incidence and has no racial or ethnic predilection. DIC has a wide range of cutaneous findings. Patients are often gravely ill and hospitalized in a critical care setting. A small subset of patients with early DIC present with cutaneous findings. The remainder of patients are first diagnosed with DIC and eventually develop cutaneous manifestations. The initial cutaneous clinical appearance is that of small petechiae that enlarge and coalesce into large macules and plaques of erythema. There may be a livedo reticularis pattern to the extremities. This fishnet-like appearance can be seen in other dermatological conditions. The petechiae quickly convert to purpuric plaques. Ulceration, necrosis, and blister formation are commonly seen in the areas of involvement. As the disease progresses, gangrene may develop in the affected areas as the blood flow to the skin is significantly decreased due to clotting of various components of the vascular system. Gangrene may lead to secondary infection. The finding of gangrene indicates a grave prognosis, and most of these patients do not survive. If DIC is treated aggressively and early, the survival rate is still only 40% to 50% at best.
DIC is considered to be a consumptive coagulopathy. The initial event that starts the reaction can be multifactorial. The most common causes of DIC are underlying malignancy (especially leukemia), severe traumatic events, sepsis, and obstetric complications. Each of these associated conditions has its own specific clinical setting. As DIC progresses, uncontrollable clotting and bleeding coexist, and patients often succumb to infection, thrombosis, or exsanguination. Thrombocytopenia is a common laboratory finding, as is an elevation of the bleeding time, prothrombin time (PT), and partial thromboplastin time (PTT). Fibrinogen is consumed, leading to an increase in fibrin degradation metabolites.
Pathogenesis: DIC may be subdivided into predominantly hemorrhagic and predominantly thrombotic types, although overlapping features of both occur in all cases. An inciting event such as trauma or infection initiates the clotting cascade in which the clotting factors are used up (or lost, in cases of severe bleeding) faster than they can be replaced. This sets off a cascade of events within the clotting system that results in consumption of all the factors used in clotting, leading to thrombosis and hemorrhage.
Histology: Examination of skin biopsies shows necrosis of the overlying epidermis and parts of the dermis. Thrombosis of the small veins and arterioles is seen, as is widespread hemorrhage. In cases of sepsis-induced DIC, evidence of the causative organism may be found in the biopsy specimen.
Treatment: Treatment requires prompt recognition of the condition and immediate supportive care. Treatment of the underlying infection is a must, and in trauma-induced cases, bleeding must be stopped and coagulation factors replaced as they are lost. The main component of therapy is treatment of the underlying cause that has precipitated the DIC event. The treatment of DIC is complicated and should be undertaken in a critical care setting. Many agents are used to help decrease thrombosis and replace lost clotting factors. A fine balance must be maintained between clotting and thrombosis. Patients with severe DIC have a poor prognosis.

Elastosis perforans serpiginosa is classified as a perforating skin disorder. This rare cutaneous eruption is believed to be caused by an abnormal expulsion of fragmented elastic fibers from the dermis. The elastic fibers penetrate the surface of the epidermis and manifest as an unusual serpiginous eruption. It has been seen as an isolated finding but also can be seen in association with many underlying conditions, including Down syndrome, Ehlers-Danlos syndrome, and Marfan syndrome.
Clinical Findings: Elastosis perforans serpiginosa is a rare cutaneous perforating skin disease. It is much more commonly seen in the young adult population, and it has a significant male predominance, with a ratio of 4 : 1 to 5 : 1. The condition has been most often reported on the neck. The eruption typically begins as small red papules with an excoriated or slightly ulcerated surface. Initially, pruritus is the main symptom. Over time, the papules coalesce into serpiginous, “wandering” eruptions. They can be annular or semicircular. The rash runs a waxing and waning course, but most cases resolve spontaneously with or without therapy. Resolution on average occurs within 6 months, but cases lasting up to 5 years have been reported in the literature. Most cases are solitary in nature. Patients with underlying Down syndrome may have only one lesion or widespread cutaneous involvement. It has been estimated that up to 1% of patients with Down syndrome will develop evidence of this rash over the course of their lifetime. Approximately 33% of cases of elastosis perforans serpiginosa are associated with an underlying disorder (see box to right ). An autosomal dominant pattern of inheritance has been described in a small number of cases, independent of any of the listed underlying conditions. The medication penicillamine has long been known to cause abnormalities of elastic fibers, and use of this medication has been shown to induce an eruption resembling elastosis perforans serpiginosa.
As the lesions progress, the epidermis ulcerates in pinpoint regions and the underlying fragmentized and abnormal elastic tissue extrudes. The areas may become more pruritic over time, and occasionally they are slightly tender. Most are asymptomatic. The appearance is most concerning for the patient and family members.
Histology: Abnormally fragmented eosinophilic elastic tissue can be appreciated on routine hematoxylin and eosin staining. Special elastic tissue stains can be used to better isolate and appreciate the elastic tissue. Examination of biopsy specimens shows an isolated area of acanthotic epidermis in which a passageway has formed. The passage begins in the superficial dermis and leads to the surface of the epidermis. This is filled with the abnormal elastic tissue, a few histiocytes, and an occasional giant cell. Early biopsies can show a cap of keratin overlying the passageway.
Pathogenesis: The cutaneous eruption is caused by the transepidermal extrusion of abnormally fragmented elastic fibers. The reason for the abnormality in the elastic fibers has yet to be determined, except in those cases induced by penicillamine. Penicillamine has been shown to disrupt proper formation of elastic tissue. The abnormally formed fibers are then extruded from the dermis.
Treatment: Many therapies have been attempted, and their use is anecdotal at best. There have been no randomized, prospective, placebo-controlled trials for the treatment of this eruption. Many destructive modalities have been attempted with varying success. Cryotherapy has the most information to support its use, but ablative carbon dioxide lasers have also been used with good results. No therapy is required, because these eruptions almost always spontaneously remit.


Abnormal accumulation of triglycerides in various tissues, including the skin, may lead to the cutaneous finding of eruptive xanthomas. The xanthomatous diseases are a diverse group of conditions with unique clinical, laboratory, and systemic findings. An abnormality in lipid and cholesterol metabolism is what links these conditions together. Fatty acids provide the body with more than 40% of its daily energy requirements. The majority of fatty acids are supplied directly by the normal diet. Proteins and carbohydrates, when present in excess, can be converted to triglycerides to be stored as a future energy source. This process makes up the remaining source of free fatty acids and triglycerides supplied to the body.
Normal metabolism of triglycerides occurs through complex biochemical pathways. Triglycerides are converted into free fatty acids, which are broken down into acetyl-coenzyme A (acetyl-CoA). Acetyl-CoA then enters the Krebs cycle to be oxidized and turned into adenosine triphosphate (ATP), one of the main forms of energy used in cellular processes.
Ingested triglycerides are broken down into free fatty acids in the lumen of the intestine by bile acids. The free fatty acids are then transported across the gut lining as chylomicrons. This process is very rapid and occurs within 6 hours after eating. The chylomicrons are absorbed by many tissues and are converted back into free fatty acids and glycerol by the enzyme lipoprotein lipase. The free fatty acids can be converted to acetyl-CoA, converted to triglyceride and stored as an energy source for later use, or used to make various phospholipids. The storage of triglycerides for future energy use is ideal, because it yields higher amounts of energy than either proteins or carbohydrates. Triglycerides can yield 9 kcal/g of energy, whereas proteins and carbohydrates produce about 4 kcal/g. This is an efficient means of storing energy. Abnormalities in the production, breakdown, or storage of triglycerides may lead to complications resulting in cutaneous and systemic findings.
Eruptive xanthomas are one of the cutaneous findings caused by an abnormality in lipid metabolism. They can be caused by various familial hyperlipoproteinemias (types I, III, and V), by medications, or as a complication of diabetes. The cutaneous findings are identical in all of these conditions. Eruptive xanthomas should not be confused with tuberoeruptive, tendinous, or planar xanthomas, because these conditions have different biochemical bases and other systemic features that are unique. Treatment of eruptive xanthomas requires a team approach including endocrinology, cardiology, and dermatology specialists.
Clinical Findings: Eruptive xanthomas, as the name implies, have a rapid eruptive onset (hours to a few days). The most common location to be involved is the buttocks, but these eruptions can be seen anywhere on the body, including the mucous membranes. They have a predilection for the extensor surfaces of the skin. They appear as yellow to slightly red-orange, dome-shaped papules with an erythematous base. Patients often complain of mild pruritus, but occasionally they describe a painful sensation when the lesions are palpated. Eruptive xanthomas are rare in both children and adults, but they are more commonly seen in adulthood. There are no racial or sexual differences in incidence.
Patients diagnosed with eruptive xanthomas that are found to be caused by a deficiency in the enzyme lipoprotein lipase are classified as having type I hyperlipoproteinemia. This is a rare form of hyperlipoproteinemia with onset in childhood. Systemic involvement is significant, with recurrent bouts of pancreatitis and hepatosplenomegaly. These patients have extremely elevated triglyceride and chylomicron levels but normal cholesterol levels. The eye may also be affected with lipemia retinalis. Lipemia retinalis can be seen only by means of a funduscopic examination. Vision is typically normal, and the patient is unaware of any eye abnormalities. The blood vessels within the eye have a creamy white color because of the excess lipid in the bloodstream. The arteries and veins are equally affected, and the only way to differentiate the two is by comparing the caliber of the vessel. The arterial light reflex is lost. The vessels appear flat, and the rest of the fundus is a uniform creamy color. Lipoprotein lipase enzyme activity can be measured, and this test is used to help diagnosis type I hyperlipoproteinemia. Eruptive xanthomas can also be seen as part of hyperlipoproteinemia type III (familial dysbetalipoproteinemia) and hyperlipoproteinemia type V. Type III has been found to be caused by a defect in the APOE gene, which encodes the apolipoprotein E protein. This protein is particularly important in clearing chylomicrons and intermediate-density lipoproteins.
Multiple medications have been implicated in the production of hypertriglyceridemia. They include isotretinoin, glucocorticoids, olanzapine, protease inhibitors (especially ritonavir), and indomethacin. Alcohol abuse can also be a cause of hypertriglyceridemia. Patients presenting with eruptive xanthomas who are taking any of these medications should have the medication discontinued or another substituted and should be reevaluated after treatment.
Diabetes is the most common cause of hypertriglyceridemia, and it probably is also the most common cause of eruptive xanthomas. Insulin is required for normal functioning of the lipoprotein lipase enzyme. Diabetic patients who are deficient in insulin have lower activity levels of lipoprotein lipase and increased levels of chylomicrons and triglycerides as a result.
On laboratory evaluation, the patient has triglyceride levels that are extremely elevated, in the range of 2000 mg/dL sometimes even surpassing the laboratory's ability to quantify it. If a sample of blood is centrifuged for a few minutes, the white to creamy-colored triglycerides will become evident and will take up a considerable amount of the specimen. On occasion, there are so many triglycerides present that the blood sample is a light creamy color even before centrifugation.
Histology: The histological findings from biopsies of early lesions of eruptive xanthomas can mimic those of granuloma annulare. Neutrophils can be evident during the formation of an eruptive xanthoma. The neutrophilic infiltrate lessens and disappears once the lesion has had time to establish itself. It is recommended that the biopsy specimen be taken from an established lesion (one that has been present for a day or two) so that more characteristic findings will be seen. Foam cells are present with a stippled cytoplasm. The number of foam cells is not as prominent as in tuberous or tendinous xanthomas. One unique finding is the presence of extracellular lipid, which is seen between bundles of collagen.
Pathogenesis: The varying conditions that can manifest with eruptive xanthomas all have unique ways of causing hypertriglyceridemia. The final common pathway in the pathogenesis of eruptive xanthomas is the presence of significantly elevated triglyceride levels.
Treatment: The main goal of therapy is to return the triglyceride level back to a normal range. Medications that can cause hypertriglyceridemia need to be discontinued. Underlying diabetes needs to be treated aggressively to get better control of glucose metabolism and insulin requirements. Those patients with familial causes need to institute dietary changes (to avoid medium-chain triglycerides), increase their activity level, and take triglyceride-lowering medications. These medications can be used for all causes of hypertriglyceridemia. The medications most commonly used to lower triglyceride levels are fenofibrate and gemfibrozil.

Erythema ab igne is an unusual rash that can develop secondary to exposure to an exogenous heat source. The name is derived from the Latin phrase meaning “redness from the fire.” It has a clinically characteristic pattern. The differential diagnosis is limited. For unknown reasons, not all persons exposed to heat sources develop the rash of erythema ab igne. Many patients develop the rash without even knowing of its existence. Reported causes have included hot water bottles, heating blankets, heaters, and computer laptops. Essentially any exogenous heat source can cause this reaction. Erythema ab igne has also been called the “roasted skin” or “toasted skin” syndrome. The exact temperature needed for the reaction to occur is unknown, and for some reason it does not occur from hot tub use, most likely because the causes of erythema ab igne are dry heat or temperatures higher than those of most hot tubs.
Clinical Findings: This condition can be seen in individuals of any race and gender. The initiating factor is an exogenous heat source that is applied to the skin. The heat source exposure is typically chronic and repetitive. Patients often notice a fine, lacy, red, reticulated macule or patch. Occasionally, no inflammatory phase is noticed, only a reticulated hyperpigmentation of the skin. Some patients do not realize that the rash is located on skin in direct approximation to a heat source. The lower back is a commonly affected area, secondary to the use of heating blankets or bottles to help treat chronic lower back pain. There have been many reports of erythema ab igne from exposures to all sorts of heat sources. Laptop computers can release a large amount of energy as infrared radiation; if someone is chronically using a laptop computer in direct approximation to their skin (e.g., anterior thighs), the rash of erythema ab igne may develop. The diagnosis is typically made by clinical examination and historical information. Patients often need to be asked whether they have been using a heating device or consistently using a laptop computer, because the correlation is not evident to them. The development of actinic keratosis or squamous cell carcinoma within the areas of erythema ab igne has rarely been reported.
Pathogenesis: Erythema ab igne is caused by the direct effects of heat on the skin. The temperature required has not been precisely defined, but the range of 43°C to 47°C seems to be most likely. In any case, there must be repeated exposure to subthermal burning temperatures. More frequent exposures and longer exposures seem to increase the risk of development of erythema ab igne. The exact mechanism by which the rash develops is unknown.
Histology: The skin may be slightly atrophic, and elastotic tissue is seen within the dermis. The rete ridges may be thinned. Some areas may show evidence of changes such as those seen in actinic keratosis. Vacuolar degeneration of the basal layer can be seen.
Treatment: The goal of therapy is to discover and remove the exogenous heat source. Once the heat source is removed, most of these rashes slowly fade away over months. Some of the hyperpigmented areas may persist, however. Use of emollient creams or Kligman's formulation has been reported. Kligman's formulation includes a retinoid, a steroid, and a skin-lightening cream. Laser therapy has also been used to decrease the pigmentary disturbance.

Erythema annulare centrifugum (EAC) is an idiopathic rash that is classified with the gyrate erythema family. It is believed to be a cutaneous reaction to many different antigenic stimuli, although no firm conclusion on the pathogenesis has been made. It has a characteristic clinical presentation that is easily recognized. The pathology of EAC is also characteristic and helps make the diagnosis by ruling out other conditions. EAC can be a marker of internal malignancy, but most cases, by far, are not associated with an underlying malignancy.
Clinical Findings: EAC often manifests insidiously. It has been reported to occur at any age and has no sexual or racial predilection. It has an unusual and peculiar morphology. The lesions start as small, pink papules that slowly expand. The patches of EAC are pink to red with a slowly expanding border. The peculiar and characteristic finding is the presence of a trailing scale. The leading edge of the rash advances and is followed by a few millimeters of fine trailing scale that continues to track the leading edge. As the rash expands outward, a central area of clearing forms. This central area is flesh colored. In tinea infections, in contrast, the scale represents the leading edge and travels in front of the expanding erythema. The main differential diagnosis is between erythema annulare centrifugum, tinea corporis, and mycosis fungoides. Potassium hydroxide (KOH) examination will rule out a dermatophyte, and a biopsy is required to differentiate EAC from mycosis fungoides.
The rash of EAC can be asymptomatic to severely pruritic. Most cases are mildly pruritic, but the most common complaint is of the unsightly appearance. The trunk is the body area most commonly involved, followed by the extremities. It is rarely seen on the face. Some areas may resolve at the same time that new areas are occurring.
Pathogenesis: The exact etiology of EAC is unknown. It is believed to be a reaction to many different antigenic stimuli. Research has suggested that EAC can be seen as a reaction pattern to an underlying tinea infection; this is thought to be a type IV hypersensitivity reaction. Many causes have been reported, including infections (fungal, bacterial, and viral) and medications, and EAC has been reported in association with many different underlying malignancies.
Histology: Biopsies of EAC lesions should be taken from the advancing border. EAC has a superficial and deep perivascular lymphocytic infiltrate. The infiltrate has a highly characteristic “coat sleeve” appearance around the vessels. The lymphocytic infiltrate is concentrated immediately around the vessels in the dermis, and the lymphocytes appear to be coating the vessel walls.
Treatment: EAC is almost always a self-limited process that spontaneously resolves. If an underlying infection is suspected, treatment and resolution of the infection has been shown to help resolve the rash of EAC. Malignancy-associated EAC is chronic in nature; it tends to resolve with treatment of the malignancy and to recur with relapses. Drug-induced EAC responds to discontinuation of the offending medication. Topical corticosteroids such as triamcinolone may be used to help decrease the erythema and pruritus.


Erythema multiforme minor, erythema multiforme major, Stevens-Johnson syndrome (SJS), and toxic epidermal necrolysis are all classified as hypersensitivity reactions, with the most common initiating event being a medication or an infection. Some authors consider these to be completely distinct entities with specific etiologies. Until that is proven, a simple way of approaching these diseases is to consider them as representing a continuum with varying degrees of mucocutaneous involvement. Erythema multiforme minor is the most likely of all these conditions to be a unique entity, because it is more commonly caused by infection (e.g., herpes simplex virus, Mycoplasma pneumoniae ). It is also more commonly seen in childhood. The other entities are much more likely to be initiated by medications. Almost all types of medications have been reported to cause these reactions, but a few classes account for most of these severe skin reactions. The classes of medications most commonly implicated are antibiotics (especially sulfa-based products), antiepileptics, allopurinol, and the nonsteroidal antiinflammatory drugs (NSAIDs).
Clinical Findings: There is no racial or ethnic predilection, and males and females are equally affected. For unknown reasons, patients with coexisting human immunodeficiency virus (HIV) infection are much more likely to develop a serious drug eruption than HIV-negative controls. The pathomechanism of this reaction is poorly understood.
Erythema multiforme minor is the most frequently seen of these eruptions. It is more common in children and young adults and can be caused by a myriad of infections and medications. Exposures to topical antigens such as urushiol in the poison ivy plant have also been reported to cause rashes resembling erythema multiforme minor. The most common cause that has been isolated is the herpes simplex virus. The rash of erythema multiforme minor can be seen in association with a coexisting herpesvirus infection or independent of the viral infection. Most episodes last for 2 to 3 weeks. A subset of patients have recurrent episodes. The rash appears acutely as a well-defined macule with a “target” appearance—a red center, a surrounding area of normal-appearing skin, and a rim of erythema that encircles the entire lesion. The peripheral rim is very well circumscribed and demarcated from the normal skin. Over a day, the macules may turn into edematous plaques. As time progresses, the center of the lesion becomes purple or dusky red. There may be only one area of involvement or hundreds in severe cases. Erythema multiforme minor affects the palms and soles; the target lesions in these areas can be very prominent and classic in appearance. The mucous membranes of the oral mucosa are involved in 20% of cases of erythema multiforme minor. Edematous pink-red plaques can be seen, as well as the more classic target lesions. If other mucous membranes are involved, the classification of erythema multiforme minor should not be used; the patient more likely has erythema multiforme major. Most cases of erythema multiforme minor self-resolve, but they do have a tendency to recur.
Erythema multiforme major has been considered by many to be the same entity as SJS. This may be true, because the pathogenesis and clinical appearance can be similar. However, subtle differences exist and warrant classifying this condition independently. Both erythema multiforme major and SJS are most often induced by medications. The mucocutaneous surfaces are affected to a significant degree. In severe cases, the mucosal membranes of the respiratory and gastrointestinal tract may also be affected. Erythema multiforme major and SJS typically begin with a nonspecific prodrome of fever and malaise. Fever is the most frequent nonmucocutaneous symptom. The rash begins insidiously as pink macules that quickly develop a dusky purple central region. The typical target-like lesion of erythema multiforme minor is usually absent in SJS but may be seen in erythema multiforme major. Erythema multiforme major is differentiated from erythema multiforme minor in that it affects a larger surface area and affects two mucous membranes.
In SJS, the dusky center of the lesion soon begins to blister, first as small vesicles and then coalescing into larger bullae. The extent and body surface area (BSA) of blistering is used to differentiate SJS from toxic epidermal necrolysis. Most authors consider blistering of 10% of the BSA and involvement of at least two mucosal surfaces to be definitive for SJS. Those cases with 10% to 30% BSA involvement have been termed SJS–toxic epidermal necrolysis overlap . Cases with greater than 30% BSA involvement are considered to represent toxic epidermal necrolysis. Light lateral pressure at the edge of a bulla or vesicle is an objective physical test that can be performed at the bedside. Spreading or an increase in size of the blister with pressure indicates separation of the epidermis from the underlying dermis and is termed Nikolsky sign .
Pathogenesis: Erythema multiforme major/SJS is believed to be a hypersensitivity reaction to certain medications. The insulting medication is thought to be metabolized into a recognizable antigen or to act as an antigen without metabolic degradation. Antibodies bind to the drug antigen and form antigen-antibody complexes that can deposit in the skin and other regions, causing an inflammatory cascade and the clinical findings.
Histology: The classic histological picture of erythema multiforme minor and major shows an acute inflammatory infiltrate along the dermal-epidermal junction. The stratum corneum is normal. There is an interface dermatitis with vacuolar degeneration of the basal cell layer. The interface dermatitis leads to necrosis and death of the basilar keratinocytes. If the necrosis spreads and coalesces, small areas of subepidermal blister formation may be seen. Erythema multiforme minor can share some features with fixed drug eruptions. In fixed drug eruptions melanophages are typically present, whereas this is not the case in erythema multiforme. Biopsy specimens of SJS and toxic epidermal necrolysis show more interface damage and blistering of the skin. The plane of separation is in the subepidermal space.
Treatment: Therapy for erythema multiforme minor and erythema multiforme major requires supportive care. The skin lesions typically self-resolve with minimal to no sequelae. Topical corticosteroids may help decrease the time to healing and decrease symptoms of pruritus. Recurrent episodes of erythema multiforme due to herpesvirus infection can be treated with chronic daily use of an antiviral agent such as acyclovir. This decreases the recurrence of herpes simplex infection and the resulting erythema multiforme reaction. Oral lesions can be treated with topical analgesics; the use of oral steroids is reserved for severe cases.
SJS can be a life-threatening condition and can progress to toxic epidermal necrolysis. For both SJS and toxic epidermal necrolysis, the cause of the reaction should be identified and withdrawn, and infections should be treated appropriately. These patients require aggressive supportive care, including wound care and fluid and electrolyte balancing. Most patients with severe involvement will benefit from the experience of a burn unit. SJS and toxic epidermal necrolysis can be treated similarly to burns, because the same technical issues are involved. There is no consensus on how to treat these two conditions with medications. The use of oral steroids early in the course of disease may help lessen the overall involvement, but steroids increase the risk of secondary infection and should not be used in patients with infection-induced disease. If used late in the course of disease, they appear not to help and only increase risk of side effects. Intravenous immunoglobulin (IVIG) has been used to treat these conditions with varying success. If used early, it may modify the disease course; if used late, it is unlikely to be of any help. The amount of BSA involved with blistering is related to the prognosis. Those with greater BSA blistering tend to fare worse than those with smaller BSA involvement.

Erythema nodosum, an idiopathic form of panniculitis, is seen in association with a wide range of inflammatory and infectious diseases. Pregnancy and use of oral contraceptives are two of the most common associations. Erythema nodosum is believed to occur as a secondary phenomenon in response to the underlying disease state. The condition typically resolves spontaneously, but in some cases it is difficult to treat. Erythema nodosum affects the anterior part of the lower legs almost exclusively.
Clinical Findings: Erythema nodosum is most commonly seen in young adult women. There is no racial predilection. The skin findings in erythema nodosum have an insidious onset. Small, tender regions begin within the dermis and develop into firm, tender dermal nodules, with the anterior lower legs almost always involved. The rash typically affects both lower legs in synchronicity. The lesions can be multifocal or solitary in nature. Most patients have multiple areas of involvement, with varying sizes of the lesions. Involvement of other areas of the body has been reported but is exceedingly uncommon. In these dermal nodules, there is a slight red or purplish discoloration to the overlying normal-appearing epidermis. If ulcerations are present, one should consider another diagnosis, and a biopsy is warranted. Although almost all cases can be diagnosed on clinical grounds, skin biopsies are required for cases that are atypical in location or have unusual features such as ulcerations, surface change, palpable purpura, or other features inconsistent with classic erythema nodosum.
The diagnosis of erythema nodosum should lead to a search for a possible underlying association. One of the most frequent causes is use of oral contraceptive pills. If the rash is thought to be related to the use of oral contraceptives, they should be discontinued, after which the lesions of erythema nodosum typically resolve. Pregnancy is another major cause of erythema nodosum. The lesions may be difficult to treat during pregnancy, but they will spontaneously resolve after delivery. Erythema nodosum may also be seen in association with sarcoid. Löfgren's syndrome is the combination of fever, erythema nodosum, and bilateral hilar adenopathy that occurs as an acute form of sarcoid. In patients with no known reason for erythema nodosum, a standard chest radiograph should be considered to evaluate for sarcoid or the possibility of an underlying fungal or atypical infection. Valley fever (coccidioidomycosis), which is caused by the fungus Coccidioides immitis, has been linked with the development of erythema nodosum. Patients presenting with erythema nodosum who have lived in or traveled to an endemic area should be evaluated for this fungal infection. Streptococcal infection and tuberculosis are two other infections that should be considered. Erythema nodosum has also been reported to occur in the inflammatory bowel diseases and in Hodgkin's lymphoma.
Histology: Erythema nodosum is a primary septal panniculitis. The inflammation is isolated primarily to the fibrous septa that are present within the subcutaneous tissue. The fibrous septa are responsible for providing a framework for the adipose tissue. No vasculitis is seen, and its presence should make one reconsider the diagnosis. The overlying dermis has a superficial and deep perivascular lymphocytic infiltrate. A characteristic finding is that of Miescher's radial granulomas, which represent multiple histiocytes surrounding a central cleft. Multinucleated giant cells are also present within the septal infiltrate.
Pathogenesis: The etiology of erythema nodosum is unknown, but it is thought to be a hypersensitivity reaction pattern to multiple unique stimuli. It is theorized that the antigenic stimulus causes the formation of antibody-antigen complexes that localize to the septal region of the adipose tissue.
Treatment: Treatment is primarily symptomatic. One must pursue the possibility of an underlying disorder. Erythema nodosum induced by medications or pregnancy resolves spontaneously once the medication is withdrawn or after delivery. Those cases associated with an underlying infection, malignancy, or inflammatory bowel disease may be longer lasting and may show a waxing and waning course. Topical corticosteroids, compression stockings, elevation, and nonsteroidal antiinflammatory agents are first-line therapies. Severe cases can be treated with a short course of prednisone. Supersaturated potassium iodide and colchicine have also been reported to be used successfully.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here
