Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

Initially described in the writings of Dr. Caleb Hillier Parry.
Exact etiology of PHA is not well understood, but is felt to have a strong autoimmune and neurogenic component.
The initial clinical manifestations include both cutaneous findings and subcutaneous atrophy.
Other forms of lipoatrophy are usually not localized to the face.
Extracutaneous manifestations are common and should be screened for in patients with PHA.
Combined medical and surgical treatment offer excellent cosmetic outcomes with very good safety.
Secondary procedures should be part of every treatment protocol.
Idiopathic progressive hemifacial atrophy (PHA), also termed Parry–Romberg syndrome, occurs in school-aged children with a slight female and no racial predominance. PHA is typically characterized by slow progressive unilateral atrophy of the skin and soft tissue of the face, with deeper involvement of the underlying muscles and osteocartilaginous structures resulting in both aesthetic and functional orofacial issues. PHA is usually restricted to one side of the face, though bilateral facial involvement has been reported. Hemiatrophy of the ipsilateral extremity (arm and/or leg) and trunk may occur, though other sites of localized scleroderma, including linear bands or plaques on the neck, trunk, and extremities are observed more commonly (approximately 5%–15%) ( Fig. 27.1 ). It is likely multifactorial in its etiopathogenesis, including a prevalent autoimmune basis provided by histologic evidence of a lymphocytic neurovasculitis (skin and brain), abnormalities on brain imaging consistent with an inflammatory and/or vasculitic process (see Fig. 27.6 ), and laboratory findings in the sera (autoantibodies) and the CSF (oligoclonal bands), supportive of this process.
 A 14-year-old girl with long-standing linear scleroderma of the head, both Parry–Romberg subtype (left hemifacial atrophy) and cutaneous en coup de sabre features (affecting her right lower face/neck). (C) She also has hemitongue atrophy, skewed palate, malocclusion, and other dental abnormalities. (D) Further examination supports hemiatrophy of right shoulder, upper back, and chest.")
 A 12-year-old girl with a 2-year history of ECDS and no neurologic complaints. (B,C) MRI of the brain demonstrated several patchy T2 FLAIR intensities throughout the right cerebral hemisphere (frontal lobe, caudate nucleus, thalamus) as well as regions of susceptibility within the right cerebral white matter.")
PHA is increasingly considered to be on the same spectrum of disorders as its companion disease, “ en coup de sabre ” (ECDS), French for “like the cut of a sword”, which describes the indentation of the scalp and forehead of these patients. Both PHA and ECDS are considered to be subtypes of localized scleroderma, linear scleroderma affecting the head, with “typical” PHA having more subcutaneous and bone atrophy (less obvious cutaneous findings) and ECDS having a hyperpigmented sclerotic cutaneous linear band with associated alopecia and linear skull depression (see Fig. 27.5 ). Many patients with craniofacial scleroderma, linear scleroderma affecting the head (face and/or scalp), have a mixture of the two conditions ( Fig. 27.2 ). When investigated, both PHA and ECDS had the same type and frequency of extracutaneous clinical manifestations, such as dental, eye, and neurologic involvement, further supporting that these conditions are on the same spectrum ( Fig. 27.1 ). Treatment often depends on the clinical assessment of the disease activity state. If cutaneous or extracutaneous manifestations appear to be advancing or show evidence of inflammation (as opposed to fibrosis), then systemic therapy with immunosuppression is warranted. This can be followed by surgical intervention when deemed in the inactive state ( Algorithm 27.1 ).
 A 13-year-old girl with typical en coup de sabre lesion affecting the forehead and scalp with resultant skull depression.")
 and scleroderma en coup de sabre (ECDS). (A) Patient with two right-sided ECDS lesions (hyperpigmentation, dermal atrophy, subcutaneous atrophy) of the forehead extending to the superior orbital rim. (B) Patient with right-sided ECDS lesion (hyperpigmentation, dermal atrophy) of the forehead extending inferiorly to the nasal tip and left-sided PRS involvement of the zygomatic and mandibular areas. (C) Patient with left-sided PRS involvement of the infraorbital, zygomatic, and mandibular areas.")
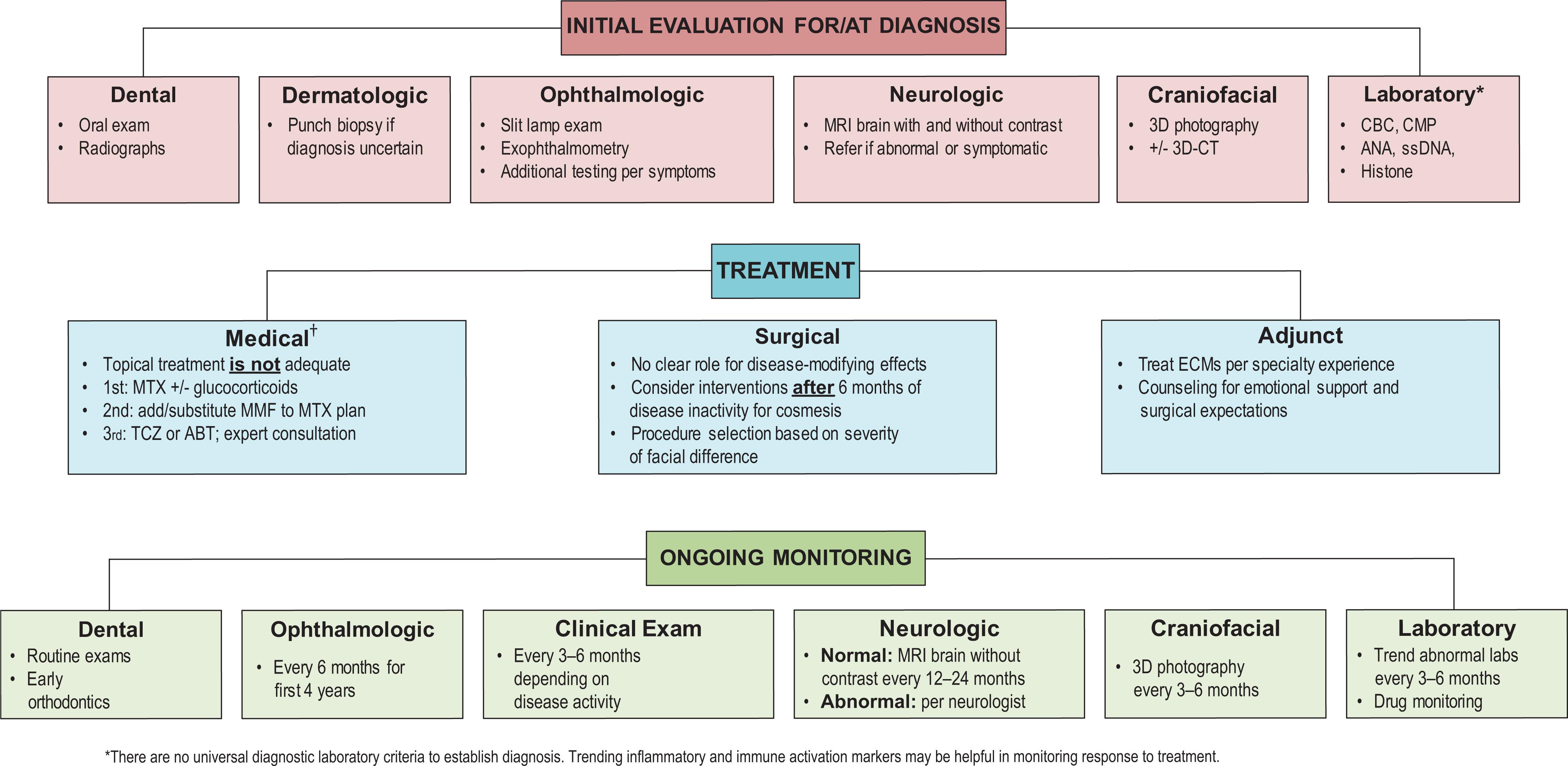
†For specific treatment regimens, see reference 35.Essential diagnostic and management algorithm for ECDS/PRS. There are no universal diagnostic laboratory or imaging criteria to establish diagnosis. Serial imaging as well as inflammation/immune activation markers may be helpful in monitoring response to treatment.
The exact etiology of PHA is not well understood but is felt to have a strong autoimmune and neurogenic component. Certain histologic findings have supported a combination of the two, which may be best described as a lymphocytic neurovasculitis. is a likely variant of the autoimmune disease localized scleroderma, specifically the subtype of linear scleroderma that affects the head. Recently the umbrella term “craniofacial scleroderma,” which incorporates both PHA and ECDS, has been proposed. Positive autoantibodies, such as anti-nuclear, anti-histone, and anti-single-stranded DNA antibodies, are demonstrated in ECDS and PHA in similar frequencies, approximately 30%–40%, and have been associated with predicting disease relapse and associating with more severe disease involvement when present.
The clinical finding of hemifacial atrophy was initially described in the writings of Dr. Caleb Hillier Parry. However, Parry’s medical writings were published posthumously on account of a stroke he suffered in 1816, which forced him to cease the practice of clinical medicine. With his daughter’s assistance, he continued his observations in writing, including observations about primary hemifacial atrophy. In 1825, three years after his death, his son Charles Parry published these writings, which included the initial description of this disorder.
In 1846, Dr. Moritz Heinrich Romberg, who had revolutionized the field of neurology in Europe by publishing the first systematic textbook on neurology, further described the clinical manifestations of hemifacial atrophy. In parallel, Thomas Addison inappropriately described circumscribed scleroderma as Addison’s keloids in 1854. This was corrected and modernized by Erasmus Wilson as morphea in 1865. Charles Fagge subsequently coined the term “ en coup de sabre” (by the strike of the sword) to describe localized scleroderma, or morphea, of the head and face in 1867, which was subsequently popularized by Jonathan Hutchinson in 1869. In 1871, Albert Eulenburg, a German neurologist, coined the term “progressive hemifacial atrophy” (PHA) and in 1945, Wartenberg published an extensive review article covering its clinical features.
The pathogenesis of PHA is basically unknown and there have been multiple suggestions for this etiology over the years. Most recently, Mulliken called this a “lymphocytic neurovasculitis” involving chronic cell-mediated vascular injury with incomplete endothelial regeneration regarding the involvement of the long branches of the trigeminal nerve.
Historically, consideration has been given to the concept that congenital hemifacial atrophy may be a type of localized scleroderma, especially when it is noted in the forehead as the “ en coup de sabre ” or “mark of the saber”. Whether these two are different entities or whether this is the same disorder in a different form (i.e., localized scleroderma) is still not entirely clear in the literature, there is increasing consensus that the two disorders lie on opposite ends of a spectrum.
In more recent years, Rogers reviewed 772 cases of PHA and there was a further definitive review in 1983 by Lewkonia and Lowry. The disorder has been shown to occur with other body asymmetries and has been described with a host of neurologic features. Many neurology articles point out that, while less common, bony deformities can be present along with the soft-tissue defect. Bilateral cases have been reported but are unusual.
In the timeline of treatment options for hemifacial atrophy, it is evident that with increased availability of modern reconstructive surgical techniques, the treatment of this disorder has gradually changed over the years. Blair, in the 1930s, and Sarnat and Neumann, in the 1950s, described the use of local flaps to augment the soft-tissue deficiency. Tube pedicles were used about the same time to transfer soft tissue into the defect from remote sites with its own blood supply and for many years this was the treatment of choice. The tube pedicles were de-epithelialized with appropriate amounts of fat and dermis used for the reconstruction. In a case noted in this chapter (from the 1960s), Barsky used abdominal tissue, which was transferred to the face using this technique.
In the 1950s, bone and cartilage grafts gained favor. Campbell, and later Converse, described the use of onlay iliac bone graft to augment areas of atrophy. Later Longacre advocated split costal bone for the same purpose. With the advent of more biocompatible metals, Kiskadden and McGregor described the use of tantalum for reconstruction of the hard-tissue defect. In the 1970s, Rees and Ashley and colleagues published their experience of treating PHA with silicone injections. Several publications followed regarding this alloplastic approach. In the 1970s, Wells and Edgerton described the use of free dermis and fat from the lower abdomen as a filler material. Fat transfer techniques have continued to be expanded upon with robust safety profile and durable outcomes, especially in those with mild–moderate facial asymmetry.
The era of free tissue transfer saw applications for the treatment of PHA. In the 1970s, Wallace described the use of an omental free flap for soft-tissue augmentation. In 1985, Jurkiewicz similarly described the use of a free, vascularized tissue for patients with PHA. By the early 1990s, a series of articles appeared by Siebert and Longaker and others using free parascapular tissue for the treatment of this disorder of severely affected patients and at the present time various modifications of this technique seem to be the standard of care.
Skin biopsies performed in patients with idiopathic PHA that had no apparent cutaneous findings demonstrated a cellular infiltrate similar to localized scleroderma: perivascular infiltrate of mononuclear cells, mostly lymphocytes and plasma cells, in the dermis, with a particular focus surrounding the dermal neurovascular bundles, termed “lymphocytic neurovasculitis” by Mulliken and colleagues. Under electron microscopy, degenerative alterations of the vascular endothelia were also documented.
Affected areas of the face with clinically evident changes of pigmentation, texture, and sclerosis tend to have more encasement and destruction of the elastic fibers and dermal appendages (hair follicles and sebaceous glands), while those with less overt superficial cutaneous changes but with more subcutaneous and soft-tissue atrophy have closely packed dermal collagen fibrils, preserved elastic fibers and hypoplastic dermal appendages. Biopsy findings, however, are variable depending on the age of the lesion as well as the depth of the sample and clinical correlation is important for correct diagnosis.
Several clinical manifestations support a neurogenic origin of PHA. The distribution of facial atrophy typically follows a dermatome of the trigeminal nerve , being unilateral in 95% of the cases and only rarely crossing the midline. Pensler et al . reported the initial distribution of atrophy among the divisions of the trigeminal nerve in 41 patients with PHA to be 35% in V1, 45% in V2, and 20% in V3, with eventual progression of disease to involve 65% in V1, 80% in V2, and 50% in V3. Neuritis of the trigeminal nerve is suggested by several patients experiencing episodes of pain in the involved area prior to the onset of tissue atrophy. An internet survey of 205 PHA patients reported 46% of responders experienced facial pain. Though most patients with PHA do not experience facial sensory, sympathetic, or parasympathetic dysfunction, some do experience peripheral facial nerve palsy, ocular motor palsy, and optic neuritis.
Another theory involving the nervous system is the hyperactivity of the sympathetic nervous system , specifically inflammation of the superior cervical ganglion, causing features of PHA. Experimental animal studies support this hypothesis. Ablation of the superior cervical ganglion of rabbits, cats, and dogs generated clinical features consistent with PHA within 30 days, such as localized alopecia, keratitis, enophthalmos, hemifacial atrophy with slight bone atrophy. Similar findings have been demonstrated in an experimental rat model after unilateral cervical sympathectomy.
In recent cohorts, approximately 30%–40% of PHA and ECDS patients are noted to have CNS involvement radiographically, though symptoms are found in less than 20% of the patients. Recent cohort studies evaluating abnormal brain imaging noted that the most common finding was ipsilateral white matter T2 hyperintense lesions and foci of susceptibility, the latter of which represent a vasculopathic or vasculitic process. Other imaging abnormalities include calcinosis, focal atrophy, and gray matter changes.
Lumbar puncture analysis of CSF reveals findings consistent with an inflammatory process, demonstrating oligoclonal bands and elevated IgG levels. Further evidence supporting inflammation of the CNS are histologic findings of brain biopsies of PHA patients, which demonstrate the same changes as seen in the ECDS form of localized scleroderma, chronic perivascular lymphocytic inflammation with some vessels showing intimal thickening and hyalinization.
As in most autoimmune diseases, infectious agents have been postulated as an etiologic agent in PHA. The most notorious suspect for both PHA and ECDS was Borrelia burgdorferi . However, further studies have not substantiated this finding. The viral infections indicated, such as Epstein–Barr virus, correlate to the typical exposure to these infectious agents in the first and second decades of life. However, they are more likely coincidental rather than etiologic factors.
The role of trauma in inducing PHA is quite controversial; however, in several patients a specific history of trauma to the affected area is elucidated, especially following tooth injury or extraction. In a self-report survey of 205 patients with PHA, 12% reported injuries they thought could be directly related to their disease onset. There have not been any standardized epidemiologic studies to verify this hypothesis.
The possibility that PHA/ECDS are related to developmental abnormalities has been incompletely described. Multiple authors have noted the concordance between cutaneous manifestations, particularly ECDS, and the lines of Blaschko; these lines delineate ectodermal migration patterning originating from the neural crest cells. Although scleroderma classically only involves mesodermal structure, the additional involvement of ectodermal derived structures, neural tissue of the brain, has prompted some authors to suggest neural crest cell abnormalities as a proximal driver of PHA/ECDS pathology.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here