Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

For those specializing in pediatric conditions, assessment of infants and children with abnormal head shapes is a common practice. Enhanced awareness through the use of social media and access to the internet has created a generation of informed parents that has resulted in an unprecedented level of advocacy. The diagnosis of an overt condition such as a cleft lip is immediate and abrupt but the nuances of metopic ridging compared with true trigonocephaly require a high level of expertise. The focus of these combined chapters is to provide a current and high-level review of craniosynostosis, its diagnosis, pathophysiology, implications, and standards of practice for successful management.
Craniosynostosis may be simply defined as the premature fusion of one or more of the cranial sutures. Because many children with craniosynostosis can be diagnosed at birth, one could wonder whether it would be classified as a failure of formation of one or more of the cranial sutures. Craniosynostosis has an incidence of between 1 in 1400 and 2100 live births, which has increased over the past two decades and is a heterogeneous group of conditions with a genetic etiology in 25% of patients.
Its impact on the affected infant will depend on the involvement of single or multiple sutures and if it is associated with an underlying syndrome or occurring in isolation. While much is known about this condition, the understanding of premature suture fusion on the effects on craniofacial growth, neurodevelopment, psychosocial well-being, and ultimately, optimal treatment paradigms continues to evolve. Goals of treatment are focused on promoting adequate brain growth, normalization of craniofacial dysmorphism, satisfactory psychosocial integration and addressing potential neurodevelopmental issues early in the child’s life.
Management of these children requires a multidisciplinary approach. Because of the complex treatments, care is best provided by centers of excellence with high volumes and access to healthcare professionals in all relevant fields. In this age of social media, family-based support groups have also become an essential and valuable resource to assist and support parents.
Historically, descriptive terms such as scaphocephaly, dolichocephaly, trigonocephaly, and turribrachycephaly were felt to be sufficient to categorize pathologic skull deformations due to craniosynostosis ( Table 25.1.2 ). It is now understood that craniosynostosis represents a heterogeneous group of conditions that differ etiologically and in clinical presentation.
| Name | Shape | Etiologic factors | Incidence |
|---|---|---|---|
| Scaphocephaly | Keel or boat-shaped | Sagittal synostosis | 1 in 2500 |
| Dolichocephaly | Long and narrow | ||
| Trigonocephaly | Triangular | Metopic synostosis | 1 in 10,000 |
| Plagiocephaly | Asymmetrical | ||
|
Unicoronal synostosis | 1 in 15,000 | |
|
Lambdoid synostosis | 1 in 30,000 to 1 in 100,000 | |
| Deformational | 1 in 3 to 1 in 7 | ||
| Turricephaly | Tower or tall | Bilateral coronal | 1 in 10,000 to 1 in 100,000 (variable) |
| Brachycephaly | Short, broad | ||
| Turribrachycephaly | Tall and short/broad | ||
| Oxycephaly | Conical or pointed | Bilateral coronal synostosis (delayed onset) | |
| Kleeblattschädel | Cloverleaf | Bicoronal and bilateral lambdoid | Rare |
Classification of craniosynostosis conditions may be by: (i) anatomic suture involved (major or minor sutures); (ii) number of affected sutures (single or multiple); (iii) syndromic or nonsyndromic; and (iv) by etiology ( Box 25.1.1 ). The majority of cases are single suture nonsyndromic but there is growing evidence to indicate an underlying genetic cause in many cases. As such, it is best to consider craniosynostotic conditions as: (1) nonsyndromic (no underlying genetic anomaly); (2) nonsyndromic with underlying genetic mutation; or (3) syndromic (in association with a constellation of clinical findings and genetic mutation).
Anatomic
Single-suture: Major
Sagittal
Metopic
Unicoronal
Lambdoid
Single-suture: Minor
Fronto-sphenoidal
Squamosal
Multiple-suture
Bicoronal
Mercedes-Benz (bilateral lambdoid and sagittal)
Kleeblattschädel
Metopic–sagittal
Any combination possible
Etiologic
Primary
Nonsyndromic – no genetic cause identified
Sagittal
Metopic
Unicoronal
Bicoronal
Lambdoid
Syndromic (> 180 identifiable syndromes)
Muenke
Saethre–Chotzen
Apert
Crouzon
Pfeiffer
Craniofrontonasal dysplasia
Nonsyndromic – genetic cause identified
TCF12-related craniosynostosis (craniosynostosis 3)
ERF-related craniosynostosis (craniosynostosis 4)
SMAD6 mutations (craniosynostosis 7)
Secondary
Biomechanical
Bone metabolic disorders
Mucopolysaccharidoses
Nutritional
Environmental
Classification based on etiology can be important. Primary craniosynostosis is due to intrinsic genetic causes acting alone or in combination with environmental factors. Secondary craniosynostosis may occur due to systemic or biomechanical causes acting on the developing suture. Secondary causes are rare but have included skeletal dysplasias (acrodysostosis, hypophosphatasia, hypophosphatemic rickets), inherited metabolic diseases such as mucopolysaccharidoses, ciliopathies, and biomechanical causes such as VP shunts. Management of these infants and children requires identification of the specific conditions for optimal treatment of the underlying condition.
The incidence of craniosynostosis has been reported to be between 1 in 1400 and 2100 live births, making this a relatively common pediatric condition. The incidence range is dependent on the subtype as certain forms of syndromic craniosynostosis and lambdoid synostosis are seen with much less frequently (up to 1 in 100,000 live births) but in several studies has been shown to be increasing over the past two decades. Syndromic craniosynostosis comprises between 12% to 31% of cases seen. Genetic anomalies are present in 25% of cases but are increasingly becoming more important in terms of understanding the functional aspects of this condition. Familial patterns are seen in 5.6% to 14.7% of cases.
Most cases of children with craniosynostosis involve a single suture and the sagittal suture is the commonest (41% to 68%) involved followed by metopic, unilateral coronal, and lambdoid. There has been a substantial increase in metopic synostosis over the past decade for reasons unknown. Symmetric forms of craniosynostosis involving the midline sutures (sagittal and metopic) are more commonly diagnosed in males and females have a higher incidence of asymmetric synostosis (coronal and lambdoid). The cause for this difference remains unknown but may be associated with the impact of circulating androgenic hormones in craniofacial development or the importance of loci on the X chromosome. In children with syndromic conditions, bicoronal and multiple suture synostosis are the most common patterns seen and the sagittal suture is much less frequently involved.
There is evidence that the incidence of nonsyndromic craniosynostosis is increasing. In 1990, the “Back to Sleep” campaign was launched in order to reduce the incidence of sudden infant death syndrome and enhanced our awareness of skull shape anomalies with the dramatic increase in deformational plagiocephaly. Possible factors related to this increase include earlier age at diagnosis, parental access to internet information sources, and improved routines by primary healthcare providers. Other factors include increased parental age, increased birth weight, and the use of antidepressants during pregnancy may also be responsible for the increase.
While over 180 syndromes have been described, the most common ones include Muenke, Saethre–Chotzen, Apert, Crouzon, and Pfeiffer syndromes. It does not appear that there has been a similar rise in the frequency of syndromic craniosynostosis which may possibly be due to earlier prenatal diagnosis (molecular genetic testing, ultrasound, prenatal MRI) leading to termination.
Geographic and ethnic variations in the incidence of craniosynostosis have been demonstrated and individuals of Caucasian and African-American descent are predominantly affected. African Americans were more likely to present with unicoronal synostosis and Caucasians were more likely to present with metopic synostosis.
Throughout history, the presence of an unusual head shape has attracted attention. The earliest references to craniosynostosis were made by Homer in the Iliad who described Thersites, a Greek warrior in the Trojan War as “bow legged, lame... with a head shaped like a sugar loaf, coming to a point and full of obscenities, teeming with rant". Plutarch, a Greek historian wrote about Pericles, the famous Athenian general as being “squill-headed” after a variety of flowering plant bulb. Both may have described oxycephaly.
In some cultures, the shape of the skull was associated with health, intelligence, and longevity. Hippocrates was among the first to allude to the pathology of cranial deformations. Galen described headache and exophthalmos as the consequences of fused sutures. Celsus, a Roman physicist who lived around the time of Christ, observed skulls without any sutures present. Oribasius (CE 325–403) noted that cranial deformities were associated with abnormalities in adjoining facial bones, such as palatal bones. He made an astute observation that pointed and asymmetric skulls were different phenotypes of the same deformity.
Andreas Vesalius was a sixteenth-century Flemish physician, and author of one of the most influential books on human anatomy, De Humani Corporis Fabrica Libri Septem ( On the Fabric of the Human Body ) published in 1543. In it, there are illustrations of skulls demonstrating suture fusion ( Fig. 25.1.1 ).
However, it was a series of German anatomists who played the most significant role in understanding the importance and physiologic properties of the cranial sutures. In the late 1790s, von Sömmerring was the first to expand beyond descriptions of cranial suture pathology and surmised the dynamic role they played in skull growth and development as well as the consequences of premature suture fusion. Subsequently, Otto, in 1830, proposed the concept of compensatory cranial expansion that occurred as the result of premature suture fusion. However, Rudolph Virchow has gained the most credit with his landmark paper published in 1851 in which he described the abnormal growth of the skull when cranial sutures were fused which was termed “Virchow’s Law”. This states that abnormal growth occurred as a result of “cessation of growth across a prematurely fused suture [in the calvarial vault]...with compensatory growth along non-fused sutures in a direction parallel to the affected suture” and was the first accurate observation and principle that would help us understand the consequences of premature suture fusion ( Fig. 25.1.2 ). Virchow originally named the condition “craniostenosis” but was convinced by Sear to use the term “craniosynostosis” which is a much better descriptive term.
Folklore has implied that several well-known historical figures including Mozart, Abraham Lincoln, and King Tutankhamen may have had craniosynostosis but there is definitive evidence that this condition was present in prehistoric times and was compatible with a reasonable level of health. Kutterer and Alt studied 76 skulls from a prehistoric population in Switzerland that included three cases of craniosynostosis. Pospíšilová and Procházková studied 745 dry skulls dated between the thirteenth to eighteenth centuries and found an incidence of lambdoid synostosis that matches today’s incidence. Astonishingly, lambdoid synostosis was described in an immature skull from the Middle Pleistocene era dated to a minimum age of 530,000 years, establishing this as the oldest evidence in human evolution of a very rare pathology ( Fig. 25.1.3 ).
The embryology of the craniofacial region is a fascinating and complex topic which is beyond the scope of this chapter but will be briefly reviewed. The skull is composed of the neurocranium (brain case) and viscerocranium (bones of the lower face). The neurocranium is further subclassified into cartilaginous and membranous components based on the method of ossification ( Fig. 25.1.4 ). The viscerocranium forms through intramembranous ossification.
Morphogenesis of the skull can be examined in two phases: embryonic (the first 8 weeks post-conception) and fetal (remaining time until birth). The two processes involve migration and condensation of the mesenchymal cells and then ossification. During the first 8 weeks of gestation, the important components that form the skull consist of the paraxial mesoderm and neural crest cells. Development of the cranial bones begins with transformation of a bilaminar to a trilaminar disc with the development of mesenchymal cells which form from epithelial cells in a process called epithelial–mesenchymal transformation (EMT). The mesenchymal cells then migrate along either side of the notochord and neural tube to form the paraxial mesoderm. Somites develop as the paraxial mesoderm forms into segments and develop in a cranial caudal progression until day 30. By the end of the fifth week, there are between 42 and 44 pairs present. The most cranial five somites and unsegmented somitomeres rostral to the first somite form the neurocranium and from ectoderm via the neural crest.
Neural crest cells are specialized, multipotential migratory cells which also form through EMT involving the epithelial cells of the neuroectoderm. They have important roles in the development of the peripheral nervous system, pigment, and endocrine cells. In the craniofacial region, these cells specifically migrate to provide the mesenchyme forming the frontal, sphenoid, squamous temporal bones and the facial bones (i.e., the viscerocranium). The parietal, petrous temporal and occipital bones form from paraxial mesoderm ( Fig. 25.1.5 ). Neural crest cell–derived tissues may be more susceptible to mutations that affect FGFR function compared to mesoderm-derived tissues. This has relevance with respect to syndromic craniosynostosis.
Once condensation of mesenchymal cells and migration of the neural crest cells is complete (end of the embryonic phase) ossification begins, as early as 7 to 8 weeks post-conception. Ossification occurs in the fetal phase and may be intramembranous (direct formation of bone) or endochondral (creation of a cartilaginous precursor with subsequent conversion into bone). Most of the bone of the craniofacial region is generated through intramembranous formation. Notable exceptions are in the ethmoid, sphenoid, petrous portion of the temporal bone and mastoid region of the occipital bone as well as inferior nasal concha which develop through endochondral bone formation. The temporal and occipital bones are compound bones containing multiple ossification centers that indicate fusion of more than one embryonic element.
Intramembranous bone formation commences with the development of ossification centers which vary from one to six, in the frontal, parietal, temporal, and occipital bones. These centers create bone spicules which radiate in a centrifugal pattern as osteoblasts differentiate from mesenchymal cells and produce osteoid (immature bone) which subsequently calcifies and forms primitive trabecular bone.
Sutures form between the calvarial bones at the site of dural reflections. These reflections consist of double folds of dura, which provides the supporting and partitioning structure of the brain. By 16 weeks of gestation, the islands of membranous bone have expanded to the location of the future sutures and represent zones which remain unossified but allow for growth through continued intramembranous bone formation. Cranial bone growth now continues at the sutures through a regulated proliferation and differentiation of osteoprogenitor cells called the osteogenic front. Rapid growth of the developing brain results in the suture expansion to form new bone through osteogenic sutural membranes. Synchondroses have similar function to sutures but form between the endochondral bones of the skull base.
The skull is a remarkable articulation of complex shapes designed to protect the vital structures for function and also to support the soft tissues that provide us with unique facial identity. The infant calvarium consists of two parietal bones, two frontal bones, two temporal bones, and the occipital bone that are joined by the four major sutures (coronal, sagittal, metopic, and lambdoid) and multiple paired minor sutures (frontosphenoidal, sphenoethmoidal, sphenoparietal, sphenosquamous, sphenopetrosal, spheno-occipital, occipitomastoid, parietosquamosal, parietomastoid, anterior and posterior intraoccipital sutures) ( Fig. 25.1.6 ).
These minor sutures align with the major sutures to form sutural arches or rings and have been classified into four groups: (1) sagittal arch consisting of the sagittal, metopic, frontonasal, and frontoethmoidal sutures; (2) coronal arch starting with the paired coronal sutures and dividing into the anterior branch (frontosphenoidal suture and sphenoethoidal synchondrosis) and posterior branch (sphenoparietal suture, sphenosquamosal suture, and sphenopetrosal synchondrosis); (3) lambdoid arch consisting of the lambdoid suture, occipitomastoid suture, and petrous–occipital and spheno-occipital synchondroses; and (4) the squamous arch comprising the parietosquamous and parietomastoid sutures with segments of the lambdoid and coronal sutures ( Fig. 25.1.7 ). While somewhat arbitrary, this classification scheme is useful in understanding the differential rates of growth of the craniofacial skeleton and the impact of premature suture fusion.
Sutures fuse in a predictable fashion. The metopic suture is the only suture to fuse during infancy between 3 and 8 months of age and does so in an ascending manner from the nasion to the anterior fontanelle. The remaining major sutures fuse between the third and fourth decades of life. Minor sutures and synchondroses fuse with cessation of facial growth, usually by late teen years ( Table 25.1.1 ).
Occasionally, islands of bone may form within sutures known as Wormian or intrasutural (ossa suturale) bones. They were first characterized by the Danish anatomist Olaus Worm in a letter to Thomas Bartholin (April 6, 1643). These can occur in any suture but are often are seen in the lambdoid suture. The os incae (Inca bone) is a single island of bone forming at the confluence of the lambdoid and sagittal suture, named due to its high incidence in Peruvian mummies. Wormian bones may be seen in multiple conditions including trisomy 21, cleidocranial dysostosis, osteogenesis imperfecta, hypothyroidism, and hypophosphatasia. They have no known clinical relevance in craniosynostosis apart from being aware of their location during surgery as possible points of dural fixation.
Occasionally, an accessory occipital suture (mendosal suture – from Latin mendosus meaning “faulty” or “incorrect”) is seen running transversely across the occipital bone. The occipital bone develops from six ossification centers. This suture separates the interparietal (planum occipitale) and supraoccipital (planum nuchale) portions of the occipital bone (representing the division between membranous and endochondral bone) and is usually fused by birth.
Intersection points of the sutures represent several important landmarks of the skull such as the bregma (point of intersection of the paired coronal and sagittal sutures), lambda (posterior intersection of the two lambdoid and sagittal sutures), pterion (the region where the frontal, parietal, temporal, and sphenoid bones join) and asterion (junction of the occipital bone, the temporal bone, and the parietal bone).
Fontanelles exist in the infant skull where three bones converge and consist of two unpaired (anterior and posterior) and two paired (anterolateral or sphenoid and posterolateral or mastoid) sutures (see Fig. 25.1.6 ). Two additional fontanelles (metopic fontanelle and sagittal fontanelle found halfway between the anterior and posterior fontanelles) can also be present but are rare. The fontanelles are non-ossified membranous regions that allow stretching and deformation of the infant cranium during delivery and as the brain expands and grows. Fontanelle closure is predictable with the posterior fontanelle closing 2–3 months after birth, sphenoidal fontanelle closing around 6 months of age, the mastoid fontanelle closes 6–18 months after birth, and the anterior fontanelle may remain patent up to 3 years of life. Bulging of the fontanelles can be an indicator or elevated intracranial pressure. Large confluent fontanelles may be present in early infancy in syndromic craniosynostosis and other conditions. The clinical finding of a small or absent anterior fontanelle is a non-specific finding and not necessarily associated with craniosynostosis.
The sutures and location of the fontanelles represent fixation points of the dura on the endocranial surface which may be susceptible to dural tears during surgery. The dura encloses the superficial venous drainage system of the brain and anatomic knowledge is essential for the pediatric craniofacial surgeon. Venous drainage from the cerebral hemispheres form into small veins in the pia mater which then form cerebral veins bridging the subarachnoid space and entering into the endothelial-lined sinuses within the dura mater ( Fig. 25.1.8 ). The superior sagittal sinus runs towards the occiput, where it receives drainage from the straight sinus. The straight sinus itself receives drainage from the inferior sagittal sinus, which courses in the falx cerebri. The inferior margin of the superior sagittal sinus divides into the right and left transverse sinus in the tentorium cerebelli and this junction point is known as the torcula. Each transverse sinus curves downward and backward as a sigmoid sinus, and is ultimately drained by each of the internal jugular veins. Venous drainage is often asymmetrical, with the superior sagittal sinus most commonly draining into the right transverse sinus, while the straight sinus usually drains into the left transverse sinus. Injuries to the venous dural sinuses during surgery may be associated with substantial morbidity or mortality. Anomalies of the venous system are commonly seen in children with syndromic craniosynostosis.
Postnatal growth of the calvarium is a rapid and evolving process that involves the response to the stimulus of the developing brain (Moss’s functional matrix theory) and a combination of sutural expansion, appositional bone growth, and remodelling. A review of a head circumference chart shows that at birth, the infant skull is 64% adult size but increases to 85% by 12 months of age then gradually slows down to 89% at 24 months of age and is nearly complete (95%) by age 7 to 8 years.
During this period, as the brain grows, the bones of the calvarium are situated on a complex membranous structure involving the dura below and periosteum above. The bones are separated as the brain expands. Tensile forces generated through brain growth are hypothesized to contribute to new bone deposition at the osteogenic fronts of the sutures and from the highly osteogenic dura. Periosteal contribution to skull growth is important but to a lesser degree compared to the dura. After 3 years of age, bone growth shifts to appositional growth with a coordinated balance between osteoblast bone apposition on the ectocranial surface and osteoclast-driven resorption on the endocranial surface allowing the bone to expand and thicken with the formation of a diploe. Appositional growth contributes to the thickness and shape of the bone whereas sutural growth results in volume expansion. Calvarial growth occurs via an increase in width through fill-in ossification of the proliferating connective tissue in the sagittal, lambdoid, parietosphenoidal and parietotemporal sutures, increase in length due to growth at the coronal sutures, and increase in height due to growth along the squamosal, occipital, and sphenoid sutures.
The impact of growth stimulus of the brain on the calvarium is highlighted by extreme examples of macrocephaly due to hydrocephalus and microcephaly due to perinatal stroke or anencephaly. Understanding the mechanisms by which sutural growth results in expansion of the cranial vault at right angles to the line of the suture (Virchow’s law) is important for recognition of the cranial dysmorphic patterns of premature suture fusion.
The role of molecular genetic analysis was discovered with MSX2 mutations detected in a family cohort in the Boston area in 1993. MSX2 is a homeobox gene that codes for a transcription factor with a major role in survival and apoptosis of neural crest cells which are important in craniofacial morphogenesis. “Boston-type” craniosynostosis is an autosomal dominant condition with substantial variable expressivity and is rarely observed outside the original cohort. Its recognition provided the opportunity for the field of molecular genetic analysis. Molecular genetic testing and assessment for chromosomal anomalies are recommended in patients with craniosynostosis.
A review of patients with craniosynostosis (n = 666) over a 13-year period treated at the Oxford craniofacial unit demonstrated that 65% of patients had nonsyndromic craniosynostosis, 2% secondary causes, 9% due to an unknown syndrome, 21% representing a clinical syndrome with an identified mutation and 3% clinically nonsyndromic but having an identified mutation. As such, 25% of these patients were diagnosed with a genetic cause for craniosynostosis. These findings are reflective of all large craniofacial units. Craniofacial phenotype alone is insufficient to establish a diagnosis of craniofacial dysostosis as significant heterogeneity exists.
There are six genes that are frequently mutated and are responsible for 75% of the cases in which a genetic mutation is present. Variants in greater than 60 genes have been identified as “craniosynostosis genes” and the list continues to expand ( Fig. 25.1.9 ). The commonest four of these genes consist of FRFR2 (Apert, Crouzon, Pfeiffer), FGFR3 (Muenke, Crouzon with acanthosis nigricans), TWIST1 (Saethre–Chotzen), and EFNB1 (craniofrontonasal dysplasia), which comprise the most recognizable cases.
 of the molecular action: arrow end = positive, transverse line end = negative, ball end = unspecified.")
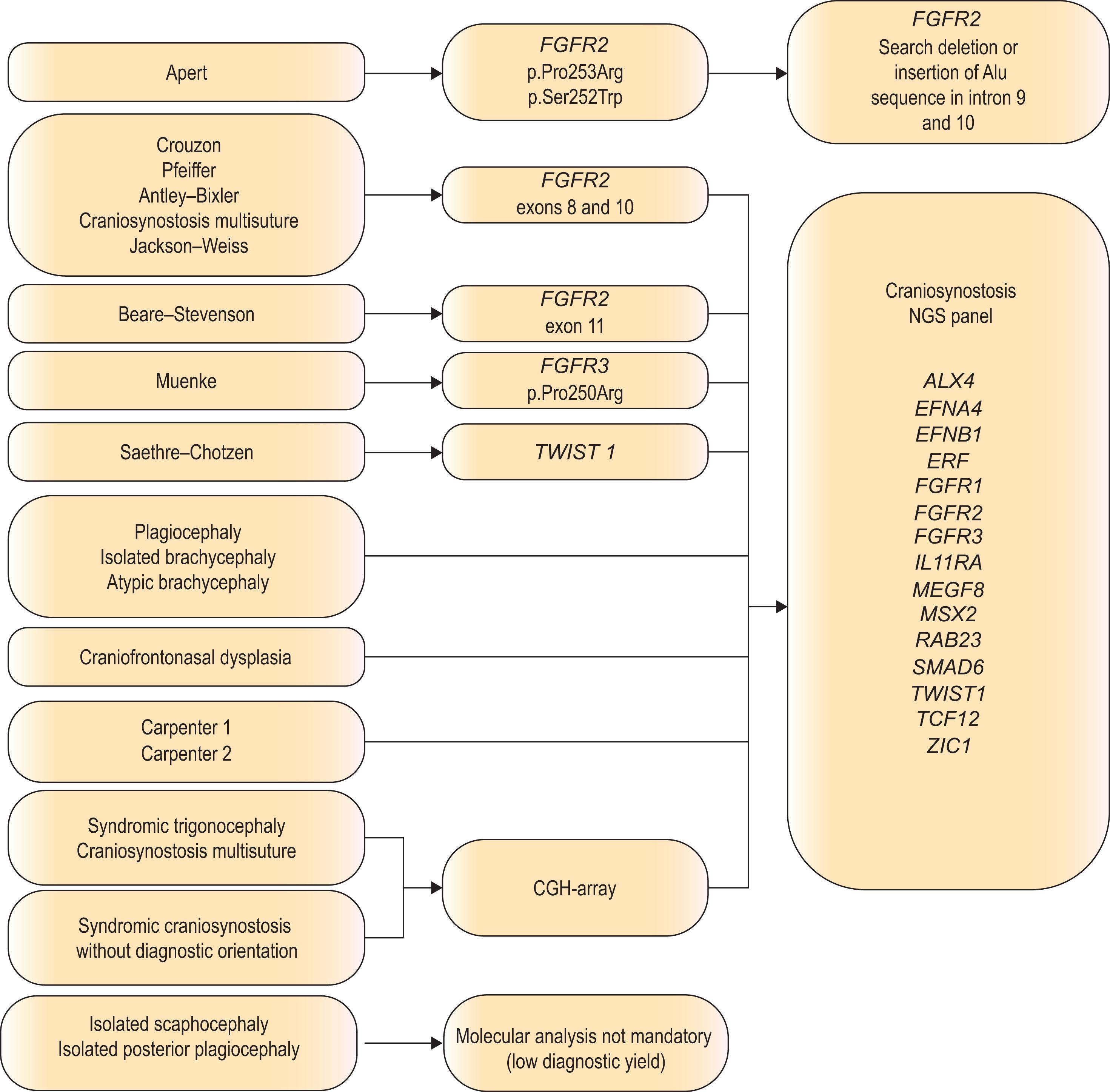
Appropriate genetic screening is recommended in all cases of craniosynostosis due to the phenotypic heterogeneity ( Algorithm 25.1.1 ). Unfortunately, genetic heterogeneity also exists. While up to 97% of children with Apert syndrome can be linked to one of two specific FGFR2 mutations (S252W or P253R), over 35 TWIST1 mutations have been identified with Saethre–Chotzen syndrome. Crouzon/Pfeiffer phenotype has been associated with at least 11 different mutations of FGFR2 . Furthermore, genetic analysis may reveal mutation rates in only 50% of Crouzon patients, 67% of Pfeiffer syndrome patients, and 71% of Saethre–Chotzen patients, suggesting that a negative genetic analysis is possible yet does not rule out the diagnosis.
Traditionally, routine genetic testing to determine a cause for synostosis depends on the clinical diagnosis. Testing children with bicoronal synostosis reveals a genetic mutation in 88% of cases but was historically much lower (<1%) for nonsyndromic midline craniosynostosis (sagittal and metopic synostosis). However, the discovery and role of TCF12 , ERF and SMAD6 has amplified these findings. ERF encodes a negative regulator of ERK1/2 which is an important signal transducer in the RAS–MAP kinase pathway and has been associated with a clinical presentation of mild Crouzon-like phenotype known as craniosynostosis 4. TCF12 encodes a partner protein of TWIST1 which is important in coronal suture development and has been associated with Muenke or SCS-like phenotype called craniosynostosis 3. This mutation accounts for up to 5% of patients originally felt to have a nonsyndromic form of craniosynostosis. The most recent addition to this group includes SMAD6 which is a protein coding gene that functions in the negative regulation of BMP and TGF-beta/activin-signaling. Damaging SMAD6 variants referred to as craniosynostosis 7 have been identified in up to 1.7% of children with sagittal synostosis, 8.1% of metopic synostosis and 20% of combined metopic–sagittal synostosis cases. SMAD6 mutations rank as the fifth most common after FGFR , TWIST1 , EFNB1 in causing craniosynostosis. Further additions to this list of monogenic causes include ZIC1 , IL11RA , ALX4 , EFNA4 , RAB23 , and MEGF8 .
There are several signaling pathways that are impacted through these gene mutations and a detailed analysis is beyond the scope of this chapter but they are covered by excellent reviews. These include FGF/Ras/ERK, bone morphogenic protein (BMP), Wnt, ephrin, Hedgehog, STAT, and retinoic signaling pathways.
A specific genetic cause can be identified in one quarter of patients with craniosynostosis and an increasing number are due to monogenic causes. These will require specific screening protocols and correctly targeted genetic testing proceeding to exome or whole genome sequencing as needed.
The causes of craniosynostosis are multifactorial and heterogeneous with monogenic and polygenic, chromosomal, environmental/teratogenic and epigenetic factors playing a role. Earlier theories included infection, metabolic conditions, biomechanical constraint, tensile forces between the cranial base and neurocranium, abnormalities in the cranial base, and primary pathology within the developing sutures. These have been replaced with advanced concepts that involve complex interactions between genetics, epigenetics, and the molecular signaling pathways important in embryogenesis and bone formation. A detailed understanding of this opaque and developing topic is beyond the scope of this chapter.
Ultimately, the pathophysiology of craniosynostosis cannot be unified by a simple theory and relates to a balance between pro-osteogenic and anti-osteogenic forces at play during suture embryogenesis. It involves the interplay of mesenchymal stem cells, neural crest cells, and their differentiated lineage with input from genetic and local tissue (dura). The theories that currently inform us about possible etiopathogenesis of craniosynostosis include: mesenchymal stem cell depletion due to increased rates of apoptosis, disturbances of homeostatic mechanisms between mesoderm and neural crest cells, imbalanced osteogenic differentiation from mesoderm and neural crest cells into osteoblasts and osteoclasts, imbalances in bone remolding with osteogenesis and bone resorption, environmental and biomechanical causes.
The majority of syndromic craniosynostoses are caused by mutations in fibroblast growth factors and their receptors (FGF/FGFR) as well as transcription factors TWIST1 and MSX2. The FGF family is made up of 22 ligands that interact with four highly conserved transmembrane receptor tyrosine kinases (FGFR1 through FGFR4) and are important for cell survival, proliferation, migration, differentiation, embryonic development, organogenesis, tissue repair/regeneration, and metabolism ( Fig. 25.1.10 ). These processes are mediated through a number of important pathways that include TGF/BMP signaling pathway, Wnt signaling pathway, Hedgehog signaling pathway and primary cilia, phosphatidylinositol-3 kinase/Akt and extracellular signal-regulated kinase (ERK)-mitogen activated protein kinase (MAPK) signaling pathways, and retinoic acid pathways, to name the main ones.
 receptor. This receptor has an extracellular ligand-binding site comprised of immunoglobulin-like domains (IgI, IgII, and IgIII), transmembrane region and divided intracellular tyrosine kinase domains, TK1 and TK2. The signaling pathway mainly operates through at least three distinct pathways. Initiation of RAS/MAPK pathways starts with formation of FRS2 complex and regulates cell proliferation and differentiation. The PI3K/AKT pathway controls cell survival and fate determination after getting initiated by FRS2 complex formation. Activation of PKC pathway started with binding of PLCγ to the activated FGFR and regulates morphogenesis and migration of cells.")
There are over 60 genes that have been felt to be causative in craniosynostosis. The majority of mutations occur as de novo mutations but may be transmitted in an autosomal dominant manner. These mutations exert their effects by perturbing physical interactions of the altered protein with its cognate protein or DNA target. A significant portion of the currently identified candidate genes are part of several interconnected functional networks. Depending on the specific affected node of these craniosynostosis-related networks, a distinct craniosynostosis syndrome or set of phenotypes arises. The FGFR family forms an interconnected network with 28 genes implicated in craniosynostosis (see Fig. 25.1.9 ). FGFR2 is critical for proper craniofacial development and involves the classic phosphatidylinositol-3 kinase/Akt and extracellular signal-regulated kinase (ERK)–mitogen activated protein kinase (MAPK) signaling cascades. The ERK–MAPK pathway is a critical regulator of craniofacial growth and suture patency. Increased FGFR2c signaling from gain-of-function mutations is associated with ERK–MAPK pathway hyperactivation and increased cellularity and dysregulated differentiation of osteoblasts in the coronal suture producing suture fusion in a mouse model of Apert syndrome.
Osteogenic differentiation from mesoderm and neural crest cells into osteoblasts and osteoclasts can be affected by several mechanisms including alterations in the Hedgehog family signaling pathway which is necessary for skeletal development. A number of conditions have been associated with genetic disorders in the Hedgehog signaling network including Greig cephalopolysyndactyly syndrome and Carpenter syndrome. Indian Hedgehog (IHH) has been shown to be present along the developing osteogenic fronts of the cranial sutures and functions to increase new bone formation, likely through upregulation of BMP2 and BMP4. FGF has been shown to promote BMP2/4 with high expression in the dura and is a main diffusible growth factor that induces suture fusion. Alternatively, Sonic Hedgehog (SHH) is present in the midline suture mesenchyme and postulated to function to maintain suture patency by increasing mesenchymal proliferation and suture mesenchymal thickness via promotion of MSX2. This balance of pro-osteogenic and anti-osteogenic mediators are important to maintain suture patency ( Fig. 25.1.11 ). IHH is expressed by calvarial osteoblasts and disruption of IHH signaling impairs osteoblastogenesis whereas overexpression of IHH may lead to suture fusion and calvarial ossification.
 is expressed by calvarial osteoblasts and disruption of IHH signaling impairs osteoblastogenesis whereas overexpression of IHH may lead to suture fusion and calvarial ossification.")
TWIST1 functions as a transcription factor in the cell nucleus and is essential for mesodermal specification and differentiation. It interacts with Runx2 which is a necessary transcription factor for osteoblast development. TWIST1 may have an extensive impact as it serves as a transcription factor to directly regulate its target genes, and also regulates other transcription factor/coregulator-mediated gene expression through interaction with other transcriptional regulators including FGF, BMP, SHH, and perhaps also TGFβ signaling. These have all shown to be relevant in osteogenesis.
In 2015, Twigg and Wilkie proposed a framework to demonstrate the intricate and temporal order of gene expression during cranial development. These pathways involve the effects of SHH on stem cell specification and migration, the WNT signaling pathway which plays vital roles in the processes of cell proliferation, differentiation, migration, and patterning, the FGFR1 , FGFR2 , and FGFR3 genes of the RAS/MAK pathway, the IHH , RAB23 , and WDR35 genes of the Indian Hedgehog pathway, in conjunction with the CYP26B1-encoding POR gene and CDC45 , which regulate the balance between osteogenic proliferation and differentiation. Prenatally, the ASXL1 and COLEC11 genes coordinate neural crest specification, migration, and maturation. Thus, mutations or changes in the expression level of any of the genes comprising this complex craniosynostosis-related network engender a distinct craniosynostosis syndrome or set of cranial phenotypes.
Epigenetics may be an important cause of craniosynostosis and can be divided into two main subgroups: biomechanical and environmental. Epigenetics is defined as heritability with or without temporary changes in gene expression without any alterations in DNA sequence. That is, some external factor can temporarily change gene expression resulting in a deformity. Biomechanical causes of craniosynostosis originally hypothesized to be the result of fetal constraint were long held but the clinical relevance is unknown. Experiments have demonstrated upregulation of Runx2 and downregulation of Noggin following cyclic loading of sutures in animal studies. This may have relevance for post-shunt suture fusion which can occur after treatment of hydrocephalus. There are a variety of environmental factors associated with craniosynostosis, which include advanced maternal and paternal age (greater than 35 years), maternal smoking, prenatal exposure to nitrosable drugs, valproic acid and phenytoin, hyperthyroidism, multiple births, birth weight >4 kg and paternal occupation (more common in agriculture, forestry, repairmen, and mechanics).
In summary, factors that are important in the pathophysiology of craniosynostosis may be likened to members of an orchestra with dizzying array of combinations of musicians playing to produce a similar end product, which is music. Structural variations may alter the dosage of one or several genes or disrupt the genomic architecture of genes and their regulatory elements within topologically associated chromatin domains. These may exert dominant effects by either haploinsufficiency, dominant negative partial loss of function, gain of function, epistatic interaction, or alteration of levels and patterns of gene expression during development. Many gene mutations involving FGRFs, TWIST1, MSX2, EFNB1 and other factors can cause craniosynostosis and exert their effect through a number of signaling pathways. These include Tgfbr1, Msx2, SKI and Gdf6 function in the Bmp pathway; Jagged 1 in the Notch pathway; EphrinA4, EphrinB1, EphA4, Dusp6, Pdgfr alpha, and FGFR1–3 in the RNT pathway; RAB23 in the Hedgehog pathway; and Axin2 of the Wnt pathway. Twist1 functions to coordinate the activities of the Bmp and RTK pathways. As such, a diverse and complex set of processes likely are responsible for craniosynostosis.
The impact that craniosynostosis has on the well-being of an infant and child will depend on the site of suture involvement, the number of sutures involved, the presence of genetic anomalies and the presence or absence of an underlying syndrome. Functional implications of craniosynostosis include possible increased intracranial pressure, the presence of intracranial anomalies, airway and ocular issues, psychosocial and neurodevelopmental concerns. The importance of an interdisciplinary approach in the management of these patients cannot be over-estimated.
Classic studies using direct intracranial pressure monitors suggested the elevated intracranial pressure (ICP) was present in up to 17% of children with single suture craniosynostosis cases and 47% of multiple suture/syndromic craniosynostosis patients. The threshold for pathologic ICP elevation in these studies was greater than 15 mmHg and has incited discussion in face of evidence that opening lumbar tap pressure in nonsynostotic children routinely ranges around 20 mmHg. This has created controversy regarding the measurement on ICP and relevance in craniosynostosis. Recent evidence suggests that between 1% and 4% of children with single suture nonsyndromic craniosynostosis may have elevated ICP using non-invasive means of measurement. The challenge arises from units who report that ICP elevation in these patients is under-reported and when measured with direct intraparenchymal monitors, have demonstrated an elevated ICP in up to 44% of untreated patients with sagittal synostosis. The normal ICP level in the growing child is unknown and the impact of intracranial hypertension on neurodevelopment continues to be debated. While untreated papilledema can result in optic atrophy and visual loss, it is unknown if this same level of ICP elevation has a detrimental impact on the developing brain. This problem is confounded by challenges of asymptomatic presentation in infants and children with confirmed papilledema and elevated ICP. It is important to recognize a low but present risk of intracranial hypertension in infants and children with single suture nonsyndromic craniosynostosis. However, it is well established that between 50% and 100% of patients with multiple sutures or syndromic craniosynostosis will have elevated ICP.
Causes of elevated ICP include craniocerebral disproportion, abnormal CSF circulation and impaired absorption, hydrocephalus, upper airway obstruction/obstructive sleep apnea and intracranial venous hypertension due to the presence of venous drainage anomalies. The concept of craniocerebral disproportion due to premature suture fusion is understandable to families. However, morphometric studies have demonstrated that in the majority of cases, intracranial volume is either normal or increased in patients with single suture craniosynostosis and, as such, craniocerebral disproportion may have a limited role as an etiologic factor.
There are several indicators of elevated ICP ( Fig. 25.1.12 ). Dilated ventricles are an indicator of hydrocephalus, which may be associated with increased ICP. Hydrocephalus may occur in craniosynostosis patients due to CSF flow obstruction as the result of posterior fossa constriction and/or impaired absorption due to venous outflow obstruction. Non-progressive ventriculomegaly is rarely seen in single suture craniosynostosis but is common in patients with syndromic diagnoses, especially Apert syndrome (90%) and may be managed conservatively. Progressive hydrocephalus requiring shunting or third ventriculostomy is more common in patients with Crouzon and Pfeiffer syndrome.
 and endocranial scalloping (B) ; more severe widespread thumbprinting (C) or copper-beaten skull with indentations of the endocranial surface (Lückenschädel skull) (D) ; small compressed tight ventricles with loss of fluid in the subarachnoid space (E) ; severe hydrocephalus (F) ; fundoscopic view showing active papilledema on the right with moderate disc elevation, indistinct margins and mild blurring of the blood vessels. (G) Image on the left is resolved papilledema – the margin is now distinct with no blurring of the blood vessels – the disc remains full and there is mild diffuse pallor; dilated veins on the scalp of an infant with sagittal synostosis due to intracranial shunting of blood through the cutaneous circulation as the result of venous hypertension suggestive of elevated ICP (H) ; Chiari I malformation with descent of the cerebellar tonsils through the foramen magnum (I) ; bulging anterior fontanelle (J) , and 3D CT scan showing bone growth around the bulging anterior fontanelle that can resemble a volcanic crater and is known as the volcano sign (K) .")
Venous anomalies can contribute to intracranial hypertension in children with syndromic craniosynostosis ( Fig. 25.1.13 ). Jugular foramen stenosis or atresia can lead to the development of collateral emissary veins, which may represent the main route for venous drainage of the cerebral venous system. Anomalies of the venous sinuses such as hypoplasia or atresia, persistent fetal sinuses and emissary veins with shunting of blood through the cutaneous circulation have been identified in up to 80% of children with syndromic craniosynostosis and were significantly associated with elevated ICP, shunted hydrocephalus, Chiari I malformation, and obstructive sleep apnea. These cases warrant preoperative imaging such as computed tomography (CT) venogram and may have relevance with respect to surgical planning ( Fig. 25.1.14 ).
 , or bilateral (B) ; absence of right transverse sinus (C) , presence of emissary veins (D) and jugular foramen stenosis (E) . A posterior skull defect (F) is associated with out-pouching of the sagittal sinus with several emissary veins seen on MR venogram (G) and during surgery (H) .")
 . Axial views demonstrating large dilated subcutaneous veins (B) . MR venogram shows an extensive subcutaneous network of veins emanating from a large emissary vein connected to the sagittal sinus (C) . Disruption of these venous channels during surgery could be associated with acute intracranial hypertension and mortality.")
Airway obstruction and obstructive sleep apnea is present in up to 75% of patients with syndromic craniosynostosis due to combinations of midface retrusion, palatal elevation, choanal atresia, and tracheal anomalies. Infants are obligate nasal breathers and, as such, airway obstruction is not uncommon in these patients. Central sleep apnea is not a common cause of airway obstruction in syndromic patients but may occur in children with a Chiari I malformation and possible brainstem compression. REM sleep-related elevations of ICP are manifested due to increased carbon dioxide levels that result in cerebral vasodilation. Sleep studies are an important part of the workup for patients with syndromic craniosynostosis.
Chiari I malformations characterized by downward displacement of the cerebellar tonsils through the foramen magnum are not a cause of elevated ICP but may occur as the result, in combination with posterior cranial fossa growth restriction and overcrowding. Its incidence may be as high as 70% in patients with Crouzon syndrome, 50% in Pfeiffer syndrome, and 100% in Kleeblattschädel deformity (see Algorithm 25.1.1 ). The presence of lambdoid craniosynostosis is associated with a higher incidence of Chiari I malformation than other patients with craniosynostosis (56% versus 0 to 10%). The symptoms of a Chiari I malformation in craniosynostosis will vary depending on the degree of herniation and can include central apnea, stridor due to vocal cord palsy, and progressive brain stem dysfunction. Decompression of the Chiari I malformation may depend on symptoms but usually follows after primary treatment of the underlying disorder (craniosynostosis, posterior fossa compression, or hydrocephalus).
Brain malformations may also be present in children with syndromic craniosynostosis and include hypoplasia or agenesis of the corpus callosum, defects of the septum pellucidum and mesial temporal abnormalities, gyral abnormalities, gray matter heterotopia, hypoplastic white matter, and megalencephaly and may be syndrome dependent.
The diagnosis of elevated ICP may not be immediately identified and should be suspected in vulnerable patients. In longstanding cases, typical symptoms in the clinical history may not be present, such as nausea, vomiting, headaches, irritability, altered mentality, failure to thrive, airway issues, sleep history, gait, swallowing, and visual symptoms. For patients with craniosynostosis, most symptoms are non-specific for craniosynostosis patients and many with elevated ICP appear clinically well. Physical findings suggestive of an elevated ICP would include tense or bulging fontanelles, widely spaced sutures and cutaneous venous dilatation. A decline in occipitofrontal head circumference was demonstrated to be a predictable indicator of the onset of intracranial hypertension. Radiographic findings include the presence of a copper-beaten skull or thumb-printing on the endocranial surface, cortical thinning, small tight ventricles and loss of extra-axial CSF or the presence of hydrocephalus (see Algorithm 25.1.1 ).
The diagnosis of elevated ICP is commonly established with fundoscopic examination for evidence of papilledema. Previous work has demonstrated the presence of papilledema was a specific (less than 98%) indicator of elevated ICP. Sensitivity, however, was age dependent. It was 100% specific in children older than 8 years of age, but its absence in younger (<8 years) children was not specific to rule out the presence of intracranial hypertension.
Due to the low sensitivity of papilledema in detecting elevated ICP, several alternative methods using non-invasive ICP measurement modalities have been described. These include optical coherence tomography, visual-evoked potential testing, transcranial Doppler ultrasound, and optic nerve sheath diameter measurement. Imaging of the fundus with spectral domain optical coherence tomography with retinal nerve fiber layer thickness is a sensitive, non-invasive technique to detect early papilledema especially when combined with clinical fundoscopic examination to assess for optic nerve pathology. Visual-evoked potentials (VEP) are also used for monitoring ICP and abnormal pattern VEPs were found to be the most sensitive indicator of elevated ICP compared to visual acuity assessment or the appearance of the optic disc. There is also evidence of electrophysiologic visual dysfunction in the majority of patients with craniosynostosis even when no clinical signs are present which makes this a very sensitive tool. Transcranial Doppler ultrasonography is another modality that has seen recent application in evaluating intracranial pressure in trauma or hypertensive causes of increased ICP. Assessment using magnetic resonance imaging (MRI) or CT have also been used to directly measure optic nerve sheath diameter as an indirect indicator of ICP elevation in patients with craniosynostosis.
The benchmark for assessment of ICP is direct intracranial monitoring using a variety of techniques (extradural, subdural, and intraparenchymal monitors). Given its invasive nature and expense of hospitalization and sedation for 24 to 48 hours, some centers utilize direct intracranial monitoring sparingly. The association of direct ICP measures and the clinical implications is not straightforward. Specifically, the “normal” ICP levels in nonaffected growing infants and children are not known, and adult values (higher than 20 mmHg) may not have clinical significance in pediatric patients. Therefore, in patients selected for direct ICP measurement, mean ICP levels with the presence of plateau waves are considered to be the most sensitive indicators of intracranial hypertension. Unchecked, elevated ICP may lead to optic atrophy and blindness. The impact of elevated ICP on neurodevelopment is less clear. Diagnosis is usually made in infancy but delayed onset of elevated ICP can be seen in late suture pansynostosis in patients with Crouzon syndrome and can also occur post corrective surgery for midline synostosis between 4 and 8 years of age. As such, screening should be performed regularly in all patients.
“Self-concept” is an idea of the self that is constructed based on how one thinks about, evaluates, or perceives oneself and on the responses of others to the self. Self-concept starts in children between ages 3 and 4 years and continues to develop through childhood and early adulthood. It includes self-esteem, self-image, self-efficacy, and self-awareness and is influenced by biological, environmental, and social interactions. Children younger than age 5 have been found to select friends based on their looks.
Anyone who has delved into the world of social media will understand the importance of the self-image. An attractive appearance is highly valued and generates opportunities for social and economic advantage and starts as early as 5 years of age. Attractiveness is gauged at a subconscious level within milliseconds when people meet. Children and young adults with a visible facial difference may have an elevated risk for psychosocial difficulties, particularly health-related quality of life. Parents of children with craniosynostosis have also demonstrated profound psychosocial impacts focused on issues around diagnosis, understanding what to expect, and justifying the need for surgery for what they perceive as a cosmetic issue.
Children and adolescents with craniofacial conditions, particularly older children with visible appearance differences (e.g., cleft lip and palate vs. cleft palate only), typically report reduced quality of life compared to both healthy adolescents and those with other chronic health conditions. A visible facial difference can have profound psychosocial implications, including altered body image, reduced health-related quality of life, and poor self-esteem. Most research suggests that a visible facial difference results in lower self-confidence and a negative self-image that might persist throughout life. Social anxiety, fear of negative social evaluation, and social avoidance are common in those with facial disfigurement. Studies of children with cleft lip have shown that affected children are at greater risk for anxiety, general unhappiness, and self-doubt in interpersonal relationships and that many affected adolescents believe their self-confidence remains affected by their disfigurement.
The impact of surgery to correct visible facial differences in adults has shown improvements in measures of personality adjustment, such as psychosis or neurosis, as well as improvements in self-concept, self-identity, self-esteem, and self-conflict. The long-term psychosocial impact of correcting craniofacial dysmorphism due to craniosynostosis is challenging to quantitate and anecdotal at best. However, aesthetic improvement of the craniofacial skeleton is the most common indication for surgical intervention in children with single suture craniosynostosis. Taking advantage of the window of opportunity when infants are too young to recall, biomechanical and osteogenic opportunities are maximized, and surgery can be performed safely within the first 12 to 14 months of life are effective levers for parents to understand. It remains to be seen what impact leaving a patient with a single suture craniosynostosis untreated will have on their psychosocial well-being. The weight of existing evidence in older patients suggests that eliminating obvious stigmata of a visible difference through surgical intervention at an age whereby no psychosocial impact has transpired would ultimately be beneficial for the health-related quality of life and well-being of the patient. Ultimately, the factors involved in decision-making around craniofacial surgery are complex and warrant further research.
Assessment of a child’s neurodevelopmental status is a complex and time-intensive process that evaluates intellectual functioning, language, processing speed, attention, executive functioning, learning and memory, visual–spatial and fine motor skills. The impact of craniosynostosis and the subsequent impact of surgery on neurodevelopment and cognition continues to be the subject of debate. Age at surgery, type of surgery, severity of the underlying condition, socioeconomic status, method of testing (global assessment or specific neuropsychological testing), age at testing, and association with cranial dysmorphology, ICP, and genetic markers are all important factors that make definitive conclusions a challenge. However, the weight of the evidence suggests that children with nonsyndromic single suture craniosynostosis are at increased risk for learning and neurodevelopmental disorders.
Studies on neurodevelopmental outcomes in children with nonsyndromic single suture craniosynostosis have demonstrated that in early development (0 to 4 years), these children are at increased risk for cognitive, language, and motor difficulties, both pre- and post-surgery. At school age (5 years onwards), intellectual functioning was within average limits but higher rates of speech and language problems were detected as well as deficits in visual–spatial skills, attention and memory associated with poorer academic performance in a minority of affected children. Assessment of the cognitive profile of patients (n = 75; ages 7 to 16 years) in Sweden with nonsyndromic craniosynostosis reported a variety of differences in these patients. Specifically, patients who had been surgically treated demonstrated a higher perceptual index than the norm. However, in patients with sagittal synostosis, the working memory and processing speed index was significantly lower, with average intelligence quotient (IQ) scores and normal intellectual ability at school age. A large cohort study tracking development of children with single suture craniosynostosis and unaffected controls (mean age of assessment at age 7.5 years) demonstrated significantly lower average IQ and math scores and slightly lower reading and spelling scores. These impacts were reported to be mild. Among all the single suture craniosynostosis types, children with metopic synostosis have the highest percentage of neurodevelopmental problems ranging from 15% to as high as 61% and sagittal synostosis the lowest (29%).
The etiology is unclear. Possible theories have implicated secondary cerebral deformation due to skull malformation. In children with sagittal synostosis, displacement of the lateral ventricles and rostrum of the corpus callosum were noted and in metopic synostosis, truncation of the posterior portion of the lateral ventricle with midline constriction of the left and right caudate nuclei were seen. However, the impact on neurodevelopment was felt to be limited due to effects of neural plasticity, compensatory processes, behavioral adaption, and environmental factors. Historically, elevated ICP and cerebral hypovascularity have been suggested as potential factors for brain impairment but a linkage seems non-specific. Previous studies have identified white matter, ventricular, and connectivity differences in patients with nonsyndromic synostosis.
However, compelling evidence to demonstrate the importance of genetics in nonsyndromic synostosis and neurodevelopmental phenotype has recently emerged. School-age children with SMAD6 mutation-influenced craniosynostosis performed significantly worse on numerical operations, performance IQ, full-scale IQ, visuomotor integration and motor coordination compared with affected children without the SMAD6 mutation. Furthermore, SMAD6-influenced craniosynostosis patients demonstrated significant behavioral pathologies including worse inhibition, hyperactivity, aggression, depression, and conduct problems. SMAD6 is an inhibitor of bone morphogenic protein signaling and functions to mediate the transforming growth factor B superfamily. These findings identify a strong genetic influence on neurodevelopmental outcomes in affected children. It is not clear the mechanisms by which these mutations translate into impact on neurodevelopment but routine genetic screening has been recommended.
Regarding syndromic craniosynostosis, there is good evidence that intellectual and developmental disability occur with greater frequency in children with syndromic craniosynostosis than in the normal population, possibly due to a combination of factors including elevated ICP, hydrocephalus, and underlying brain malformations. Although IQ studies are inadequate as an indicator of neurocognition, IQ scores in children with syndromic craniosynostosis are significantly lower than normative population scores (mean full scale IQ 83.1 vs. 100).
The impact of surgery on neurodevelopment in children with single suture nonsyndromic craniosynostosis continues to draw debate. Intuitively, increasing space for a growing brain makes sense but direct correlation between ICP and brain development is lacking. Patients with unoperated sagittal synostosis have shown worse performance with regard to attention, processing, memory, learning, and language compared with normal populations. There is some evidence that earlier surgery and the type of procedure is beneficial for patients with sagittal synostosis. Specifically, in a comparison of only patients treated before 6 months, those patients undergoing whole-vault cranial reshaping demonstrated superior performance relative to those patients who had strip craniectomy based on IQ (P <0.05), verbal IQ (P <0.01), word reading (P <0.05), and reading comprehension (P <0.05). There was no evidence that later surgery is of benefit. In a recent robust systematic review on the impact of surgery timing for craniosynostosis on neurodevelopmental outcomes, Mandela and colleagues stated that it was difficult to draw conclusions due to the presence of multiple confounding variables but some inconclusive evidence suggests that earlier (<6 months of age) was better for sagittal synostosis. In their assessment of 10 studies, 5 demonstrated a beneficial effect on neurodevelopmental outcome and 5 did not. No study assessed the effect of anesthesia and only 1 study assessed socioeconomic factors. Until an adequately powered prospective study can be performed, the subject of surgical intervention and neurodevelopmental benefit continues to evolve. Definitive conclusions cannot be made at this time. The role of neurodevelopmental screening and early diagnosis for referral remains essential in the management of these children.
Ophthalmic issues are common in children with craniosynostosis. Visual compromise may occur due to a number of reasons including amblyopia, strabismus, astigmatism, optic atrophy due to elevated ICP, and exposure keratopathy. Extreme presentations of exorbitism and ocular globe proptosis resulting from growth restrictions of the bony orbit may occur in children in syndromic craniosynostosis such as Pfeiffer and Crouzon syndrome. Chronic corneal exposure and lack of adequate tear film protection can lead to a range of manifestations including superficial punctate keratitis, corneal ulceration, corneal scarring, and in extreme cases, endophthalmitis and eyeball perforation. When the bony orbit is shallow, the eyelids may be at a mechanical disadvantage and, in rare cases, unable to provide adequate eye closure to protect the cornea.
Amblyopia is the most common cause of visual impairment in children with craniosynostosis and occurs with deprivation of vision in one or both eyes resulting in under-stimulation of the visual cortex. Amblyopia may occur due to any etiology that compromises the visual axis, such as strabismus, refractive errors, or corneal scarring.
Strabismus occurs in 39% to 76% of patients with craniosynostosis and may be commonly seen in up to 50% of unicoronal craniosynostosis (UCS) cases. While the contribution of the shape of the bony orbit to extra-ocular muscle dysfunction is not clear, evidence demonstrates that infants with UCS undergoing minimal access endoscopic strip craniectomy correction with helmeting had less severe overelevation in adduction, amblyopia, extremes of astigmatism and less need for strabismus surgery compared to those who had an open approach with frontal–orbital reshaping. Earlier surgical intervention or differences in anatomical changes were hypothesized to play a role.
Optic neuropathy due to chronically elevated ICP occurs with papilledema, neuronal death, optic nerve atrophy, and permanent visual loss. Stasis of axoplasmic flow at the optic nerve head leads to intraneuronal ischemia causing neuronal death. This may manifest as loss of visual acuity or change in visual fields.
Papilledema occurs as the result of elevated ICP transmitting across the lamina cribrosa and optic nerve sheath to induce axoplasmic flow stasis and cause swelling in the optic nerve head. The presence of papilledema was found to be 98% specific as an indicator for elevated ICP. The sensitivity was age-dependent and was 22% in children less than 8 years of age. Papilledema may not occur in young children with elevated ICP which is concerning given its use as a screening tool. Papilledema may not occur in cases where extensive optic nerve atrophy is present because the destroyed axons no longer have capacity to swell. Therefore, a diagnosis of elevated ICP therefore cannot be ruled out on the basis of papilledema not being apparent. True papilledema arising from elevated ICP must be differentiated from pseudo-papilledema which most commonly arise from optic disc drusen. Drusen consist of cytoplasmic material accumulations within the optic disc that are commonly located deeply in children, in contrast to the superficial location in adults which make them easier to diagnose. Using fundoscopy, this may appear as an elevated optic disc that does not obscure underlying retinal vessels, in contrast to the vessel obscuration that commonly occurs with true papilledema.
It is recommended that comprehensive ophthalmologic examination be performed biannually until 7–9 years of age and then annually yearly for children with syndromic craniosynostosis. For children with nonsyndromic craniosynostosis, these examinations should be performed annually until 7–9 years and as needed thereafter. Evaluations should include assessment of visual acuity, ocular alignment, refractive error, and examination of the cornea and optic nerve; routine fundoscopic exam is particularly important to evaluate for papilledema in these patients.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here