Physical Address
304 North Cardinal St.
Dorchester Center, MA 02124

Bone is a highly specialised tissue ( E-Fig. 22.1 H ), composed of a particular type of collagen embedded in a ground substance matrix, osteoid . Osteoid becomes mineralised by the deposition of calcium salts in the form of hydroxyapatite . The osteoid is synthesised by osteoblasts, which, in their resting state, are inactive, spindle-shaped cells lying unobtrusively on bone surfaces. When there is a need to synthesise more osteoid, osteoblasts become cuboidal and actively synthesise protein ( E-Fig. 22.2 H ). Bone is constantly being remodelled through the removal of old bone by resorptive cells called osteoclasts ( E-Fig. 22.3 H ) and deposition of new osteoid by osteoblasts. Thus, the bulk and architectural arrangement of bone can be modified in response to changing functional demands and stresses. As well as its structural role, the calcified bone also provides a large calcium pool from which calcium can be withdrawn (by resorptive activity of osteoclasts) to maintain serum calcium homeostasis.
Bone fracture is a common and important result of trauma. Where there is an underlying bone abnormality, for example osteoporosis, osteomalacia, Paget’s disease or metastatic tumour, the trauma or extra stress required to produce bone fracture may be minimal. This is called pathological fracture . The healing of a bone fracture is briefly outlined in Fig. 3.11 as an example of a specialised form of tissue repair.
Infections of the bone are now comparatively uncommon, although bacterial osteomyelitis, both pyogenic and tuberculous, were formerly important crippling diseases. Acute osteomyelitis ( E-Fig. 22.4 G ) is caused by pyogenic bacteria such as Staphylococcus aureus ( Fig. 22.1 ). This usually occurs in infants and young children, the bacteria gaining access to the marrow cavity via the bloodstream. It may also follow penetrating trauma in people of any age, for example after compound (open) fracture. Pathologically, acute osteomyelitis represents abscess formation in the medullary space of the bone, but the course of the disease in bone is complicated by two factors. First, increased pressure in the confined space causes infarction of further large areas of bone. Second, masses of dead bone (sequestra) behave as foreign bodies, inhibiting normal repair processes and providing a haven for bacteria that is inaccessible to body defence mechanisms. Chronic osteomyelitis ( E-Fig. 22.5 G ) may follow if treatment is delayed or inadequate. Tuberculous osteomyelitis is illustrated in Fig. 5.7 .
 (HP); (B) necrotic bone (HP).")
Osteoporosis (osteopenia) is a condition in which total bone mass is decreased due to reduction in the number or size of bone trabeculae (usually both) and thinning of cortical bone. Despite this, the bone otherwise appears structurally normal. In contrast, osteomalacia is a disease in which osteoid fails to undergo normal mineralisation, usually because of deficiency of vitamin D, although other causes of total body calcium depletion may also be responsible. Rickets is the childhood equivalent of osteomalacia. The appearances of normal bone, osteoporosis and osteomalacia are compared in Fig. 22.2 .
 (MP). (A) Normal bone; (B) osteoporosis; (C) osteomalacia.")
Paget’s disease of bone ( Fig. 22.3 ) is a condition of unknown aetiology that occurs in the elderly. It is characterised by haphazard and inappropriate osteoclastic erosion of formed bone and concurrent, haphazard osteoblastic deposition of new bone. Hyperparathyroidism , due to excessive secretion of parathyroid hormone by a parathyroid adenoma or parathyroid hyperplasia, may cause rather similar haphazard osteoclastic erosion of bone.
L lacunae M marrow space N necrotic bone Ob osteoblasts Ost osteoid
Loss of bone mineral density is a common clinical problem that gives rise to considerable morbidity and mortality owing to the occurrence of pathological fractures through the weakened bones. Such fractures occur most often in post-menopausal women and typical sites of fracture include vertebrae, femoral neck and distal radius (the so-called Colles’ type fracture ). Such injuries can result in prolonged periods of reduced mobility, exacerbating the underlying problem of reduced bone mass.
Interventions focus on trying to reduce the risk of such fractures, as well as aiming to avoid subsequent immobility if fractures do occur. Prevention of osteoporosis requires a range of interventions, aimed at maximising bone mass by promoting weight-bearing exercise and good nutrition. In higher-risk patient groups, supplementation with calcium and vitamin D may be encouraged. Drugs such as bisphosphonates act by preventing bone resorption by osteoclasts, and are widely used in clinical management of osteoporosis and other metabolic bone disorders.
. (A) Active osteolytic lesion (undecalcified resin section, Goldner’s trichrome); (B) inactive sclerotic lesion (decalcified paraffin section, H&E).")
Bone is a frequent and important site of haematogenous metastatic spread of malignant epithelial tumours, particularly carcinomas of bronchus, breast, kidney, thyroid and prostate (see Fig. 16.14 ). Metastatic tumour deposits usually destroy bone trabeculae, although carcinoma of the prostate (see Fig. 19.10 ) sometimes stimulates excessive new bone formation, resulting in osteosclerotic rather than the more usual osteolytic deposits.
Primary tumours of bone are much less common. These may arise from any of the cell types found in bone. The essential features of the more important of these tumours are given in Table 22.1 .
Osteoid osteoma is a benign but painful tumour of bone, which is illustrated in Fig. 22.4 .
 Osteoid osteoma in medullary cavity (LP); (B) histology of edge of lesion (MP).")
Osteogenic sarcoma or osteosarcoma is the most common malignant primary tumour of bone ( Fig. 22.5 ). This occurs mainly in children and adolescents.
 (HP).")
Chondromas ( Fig. 22.6 ) are benign tumours of hyaline cartilage, which may arise either on the surface of bone or within the medullary cavity where they are known as enchondromas . These may rarely undergo malignant transformation.
.")
Chondrosarcomas ( Fig. 22.7 ) are malignant cartilage tumours that mainly arise in middle-aged and elderly individuals.
.")
| Name of tumour | Main pattern of cellular differentiation | Age and sex incidence | Common sites | Behaviour |
|---|---|---|---|---|
| Osteoid osteoma ( Fig. 22.4 ) | Osteoblast | Adolescents, M>F | Long bones, lower limb | Benign, painful, osteosclerotic |
| Osteosarcoma (Osteogenic sarcoma) ( Fig. 22.5 ) | Primitive osteoblast | Mainly adolescent Elderly, M>F |
Around knee Sites of Paget’s |
Highly malignant, metastasis to lungs |
| Chondroma (Enchondroma) ( Fig. 22.6 ) | Chondrocyte | Common, any age | Hands and feet, long bones | Benign, very rare malignant change |
| Chondrosarcoma ( Fig. 22.7 ) | Chondrocyte | Middle aged/elderly, M>F | Pelvis, ribs, vertebrae | Malignant, local spread and metastasis |
| Non-ossifying fibroma | Fibroblast | Mainly children and young adults | Long bones of lower limbs | Benign, osteolytic, may be multiple |
| Chondromyxoid fibroma | Uncertain | Young adults | Long bones, mainly tibia | Benign, osteolytic, may be painful |
| Giant cell tumour | Osteoclast | 20–40 years, M>F | Long bones, mainly around knee | Benign, may recur locally |
| Chordoma | Notochord tissue | Middle aged/elderly, M>F | Sacrum, skull base | Locally aggressive bone destruction |
| Ewing’s tumour ( Fig. 7.6 B) | Uncertain | Children/adolescents, M>F | Shaft of long bones | Highly malignant, early metastases |
As discussed in Ch. 16 , malignant tumours may also arise from the haematolymphoid cells of the bone marrow, for example myeloma, lymphoma, leukaemia, etc.
In addition to true bony neoplasms, there is a miscellaneous group of tumour-like lesions that are also found in bone. The main features of these lesions are presented in Table 22.2 . The cartilage-capped exostosis or osteochondroma is the most common of these and is illustrated in Fig. 22.8 .
M mosaic lines Ob osteoblast Oc osteoclast Ost osteoid
Primary malignant bone tumours occur mainly in children and adolescents and, traditionally, the prognosis has been extremely poor. In the past, surgical excision of the primary tumour mass was the mainstay of treatment and this normally required amputation of the affected part. With the development of newer chemotherapeutic agents, treatment and outcome in these cases has altered considerably.
Following initial biopsy for diagnosis and classification of the tumour, most patients are now treated with chemotherapy in the first instance and the tumour response is monitored closely by regular radiological imaging. After this, complete surgical excision of the residual tumour is undertaken. Amputation of limbs is avoided whenever possible by the use of new bone and/or joint prostheses, which can be surgically inserted to replace the affected parts of the skeleton, leaving surrounding tissues intact. This strategy of initial treatment with systemic chemotherapy has dramatically improved the outlook for children with malignant bone tumours.
A atypical mitosis B binucleate tumour cells C chondrocytes H hyaline matrix MO mineralised osteoid Ob osteoblast Ost osteoid T tumour
| Name of lesion | Age and sex | Common sites | Behaviour |
|---|---|---|---|
| Osteochondroma (exostosis) | Children/adolescents, M>F | Upper tibia, lower fibula | Non-neoplastic outgrowth of bone and cartilage, possibly derived from growth plate. |
| Aneurysmal bone cyst | Children/adolescents, M>F | Long bones, spine | Reactive, cause unknown. Osteolytic, risk of fracture at site. |
| Fibrous dysplasia | Children/adolescents, M>F | Long bones, facial bones | Developmental abnormality, 20% involve multiple bones. Osteolytic, risk of fracture. |
| ‘Brown tumour’ of hyperparathyroidism | Adults, M>F | Anywhere | Osteolytic, often multiple. Resemble giant cell tumour. Treat hyperparathyroidism. |
| Disorder | Main features | Figure |
|---|---|---|
| Non-neoplastic bone disorders | ||
| Fracture | Common following trauma. Specialised form of repair with callus. Pathological fracture occurs through bone weakened by disease. | 3.11 |
| Osteomyelitis | Pyogenic organisms such as Staphylococcus aureus , TB now rare. Haematogenous spread or after penetrating injury, e.g. open fracture. Sequestrum of dead bone inhibits resolution. | 22.1 |
| Osteoporosis | Reduction in bone mass. Qualitatively normal bone mineralisation. Common cause of pathological fracture in postmenopausal women. | 22.2B |
| Osteomalacia/rickets | Incomplete mineralisation of normal sized trabeculae. Due to vitamin D deficiency. Called rickets in children and associated with deformity. | 22.2C |
| Paget’s disease of bone | Uncontrolled osteoblastic and osteoclastic activity. Thickened but weakened bone. Mosaic cement lines. Risk secondary osteosarcoma. | 22.3 |
| Benign and tumour-like lesions of bone | ||
| Osteochondroma (exostosis) | Non-neoplastic outgrowth of bone with cartilage cap at epiphysis. Hereditary form with multiple lesions. Small risk of chondrosarcoma. | 22.8 |
| Osteoid osteoma | Benign bone-forming tumour in medullary cavity of long bones. Central nidus. Typically painful and relieved by aspirin. | 22.4 |
| Chondroma (enchondroma) | Benign cartilage-forming tumour. Enchondroma in medullary cavity. Rare syndromes with multiple lesions, e.g. Ollier’s disease. | 22.6 |
| Malignant bone tumours | ||
| Metastatic carcinoma | Commonest form of malignant tumour in bone. Typically from lung, breast, prostatic, renal and thyroid carcinomas. Usually lytic on X-ray examination. | 16.14 |
| Osteosarcoma | Malignant bone-forming tumour. Children/adolescents or secondary to Paget’s in elderly. Long bones, early haematogenous spread. | 22.5 |
| Chondrosarcoma | Malignant cartilage-forming tumour. Axial skeleton, usually older adults. | 22.7 |
| Ewing’s sarcoma | Small round blue cell tumour. Usually children, typically shaft of long bones. Highly aggressive behaviour. | 7.6B |
| Joint disease | ||
| Osteoarthritis | Common degenerative disease. Fibrillation of hyaline cartilage, eburnation, subchondral cysts and osteophyte formation. | 22.9 |
| Gout | Crystal arthropathy. Deposition of uric acid crystals in synovial fluid and/or forming tophi. Needle-shaped, strong negative birefringence. | 22.10 |
| Pseudogout | Crystal arthropathy. Deposition of calcium pyrophosphate crystals in large joints. Rhomboid crystals, weak positive birefringence. | |
| Septic arthritis | Pyogenic and tuberculous types. Usually single acutely painful joint. Needs urgent antibiotic treatment to limit joint damage. | |
| Rheumatoid arthritis | Systemic inflammatory disease with symmetrical small joint polyarthropathy. Inflamed synovium, sometimes rheumatoid nodules. | 22.11 |
| Benign and reactive soft tissue lesions | ||
| Traumatic neuroma (amputation neuroma) | Reactive proliferation of small nerve branches in scar following transaction of peripheral nerve. Morton’s neuroma on foot similar. | 22.12 |
| Ganglion | Reactive cystic lesion, usually hands or wrists. Myxoid degeneration and cysts associated with tendon sheath or near joint. | 22.13 |
| Giant cell tumour of tendon sheath (nodular tenosynovitis) | Common, typically fingers. Localised firm nodule. Mixture of giant cells and mononuclear cells with haemosiderin and foamy histiocytes. | 22.14 |
| Pigmented villonodular synovitis | Similar to giant cell tumour but diffusely affects single joint. Commonly knee, often locally recurrent after treatment. | Like 22.14 |
| Lipoma | Common benign tumour of fat. Soft swelling, usually in subcutaneous tissues. Mature adipose tissue. | 22.15A |
| Angiolipoma | Similar to lipoma but often painful due to small, capillary vessels with fibrin thrombi. Often multiple. | 22.15B |
| Leiomyoma | Benign smooth muscle tumour. In skin, well-defined and derived from vessels (angioleiomyoma) or with irregular edge (from pilar muscle). | 7.12 17.10 |
| Haemangioma | Benign proliferation of blood vessels. Common in skin. In liver, association with hormonal contraceptives. Capillary or cavernous sized channels. | 11.8 |
| Schwannoma | Benign tumour of Schwann cells. Encapsulated. Spindle-shaped cells with cellular Antoni A (nuclear palisading) and myxoid Antoni B areas. | 23.15 |
| Neurofibroma | Benign tumour of peripheral nerves. Ill-defined edges. Spindle cells. Multiple in von Recklinghausen’s disease (neurofibromatosis). | 23.16 |
| Rhabdomyoma | Rare benign tumour of striated muscle. Cardiac rhabdomyomas typically occur in young children. | |
| Malignant soft tissue tumours | ||
| Liposarcoma | Malignant fatty tumour. Adults, deep soft tissue sites. Lipoblasts typical. Various types. Molecular methods helpful. | 22.16 |
| Leiomyosarcoma | Malignant tumour of smooth muscle. Pleomorphic spindle-shaped cells with eosinophilic cytoplasm. | 7.6 |
| Angiosarcoma | Malignant tumour forming vessels. Aggressive behaviour. May complicate chronic lymphoedema or follow irradiation. | 11.12 |
| Malignant peripheral nerve sheath tumour | Malignant tumour derived from peripheral nerves. Association with neurofibromatosis. | |
| Fibrosarcoma | Malignant proliferation of fibroblasts. Typically, fascicles of spindle cells with ‘herringbone’ architecture. | |
| Synovial sarcoma | Sarcoma of uncertain histogenesis. Typical combination of spindle cells and epithelioid areas (biphasic). Cytogenetics shows t(X;18). | |
| Undifferentiated pleomorphic sarcoma | Pleomorphic, high grade tumour with no clear evidence of differentiation. Older adults, aggressive behaviour. | 22.17 |
.")
The three most important disorders of joints are osteoarthritis , gout and rheumatoid arthritis . Osteoarthritis ( Fig. 22.9 ) is the name given to the wear and tear degenerative changes that occur in some joints with increasing age. Gout is an inflammatory arthropathy caused by the deposition of urate crystals within the joints and soft tissues (illustrated in Fig. 22.10 ). Rheumatoid arthritis is a systemic chronic inflammatory disorder of autoimmune origin characterised by synovitis and arthritis ( Fig. 22.11 ).
Joint pain is a fairly common symptom, but one that requires careful assessment. In particular, the rapid onset of severe pain in a single joint should be investigated immediately.
Gout can present in this way, as can another crystal arthritis called pseudogout , but acute pyogenic joint infection (septic arthritis) is a very important differential diagnosis since this requires urgent antibiotic treatment to prevent destruction of the joint. Other forms of arthritis, such as rheumatoid disease, tend to affect multiple joints (polyarthropathy) , but the initial presentation is sometimes with involvement of a single joint.
Clinical examination and initial investigations such as a full blood count should be followed by aspiration of synovial fluid from the joint. Immediate examination of this specimen can be helpful in guiding treatment. Fluid can also be submitted for microbiological culture in cases of suspected joint sepsis.
Normal synovial fluid is virtually acellular but, in acute pyogenic infection, it may be transformed into pus, consisting of numerous neutrophil polymorphs. Some neutrophils are also seen in cases of acute crystal arthropathy but here, polarisation reveals the presence of crystals. As illustrated above, uric acid forms fine, needle-shaped crystals that show strong negative birefringence . This term refers to the physical property of rotating polarised light in a particular direction as it passes through the specimen.
In contrast, pseudogout is caused by precipitation of calcium pyrophosphate crystals that are rhomboid in shape and show positive birefringence. This disorder tends to affect large joints such as the knees, often in elderly patients who are being treated with diuretic drugs.
A articular surface B bone CC cartilage cap D cystic degeneration GC giant cell U urate crystals V vessels
C cartilage F fibrous tissue GC giant cell N necrosis NB nerve branches P pannus PH palisading histiocytes
Many benign tumours and reactive tumour-like lesions commonly occur in soft tissue. Malignant soft tissue tumours (sarcomas) are rare and their diagnosis and classification form a highly specialist area of pathology. Here, we will illustrate some of the reactive lesions and benign tumours frequently encountered in clinical practice and also review the broad concepts of sarcoma pathology by consideration of a few specific tumour types.
Some soft tissue masses do not satisfy the usual criteria to be classified as true benign neoplasms and are considered to be reactive rather than neoplastic . These lesions may arise following a minor injury or in areas exposed to repetitive trauma, but such a history is not always obtained. The line between benign soft tissue tumours and reactive tumour-like lesions is not always clear and, in recent times, molecular evidence of clonality has begun to emerge for some of the entities conventionally considered to be reactive.
It is of great importance to note that some reactive soft tissue lesions may be very rapidly growing, causing clinical suspicion of malignancy. Such lesions may also appear alarming histologically, showing active proliferation with numerous mitotic figures. This possibility must always be considered before suggesting a diagnosis of soft tissue sarcoma.
A large number of soft tissue masses fall into this broad group, but the majority are encountered only rarely in clinical practice. A few of the more common lesions are considered here. The traumatic or amputation neuroma is a reactive mass that may form following damage to a peripheral nerve ( Fig. 22.12 ). A ganglion is a fairly common lesion that is probably related to repeated minor damage around sites of excessive tendon and joint use, e.g. around the wrists and hands of pianists and typists ( Fig. 22.13 ). Giant cell tumour of tendon sheath (also known as nodular tenosynovitis ) also tends to occur around the hands and wrists ( Fig. 22.14 ). As the name suggests, the nature of this lesion has been the subject of considerable debate and there is increasing evidence that it may be a true benign neoplasm.
Some reactive processes in soft tissue appear extremely alarming, both clinically and histologically, and may be described as pseudosarcomas . One example is nodular fasciitis . This typically presents as a very rapidly growing soft tissue swelling, appearing over the course of a few weeks. On close questioning, there may be a history of preceding trauma. The mass tends to be situated in the superficial fascia and usually has ill-defined margins (unlike most benign tumours, which tend to be encapsulated). Histologically, this is a proliferation of spindle cells, often with very numerous mitotic figures. There is a high risk of over-diagnosing malignancy in this setting and great care is required to recognise that this is a reactive process, rather than a sarcoma.
C cystic space FM foamy macrophages GC giant cells H haemosiderin M myxoid change T fibrin thrombus W fibrous wall
 Early changes (MP); (B) established lesion (LP).")
 H&E (MP); (B) as above, viewed using plane polarised light.")
 Synovium (MP); (B) articular cartilage (MP); (C) rheumatoid skin nodule (LP); (D) rheumatoid skin nodule (HP).")

.")
. (A) LP; (C) HP.")
One of the commonest of all soft tissue lesions is the lipoma , a benign tumour that resembles normal adipose tissue ( Fig. 22.15 ). Other benign soft tissue tumours may show patterns of differentiation resembling smooth muscle ( leiomyoma , see Figs 17.10 and 7.12), blood vessels ( haemangioma , see Fig. 11.8 , peripheral nerves ( Schwannoma , see Fig. 23.15 and neurofibroma , see Fig. 23.16 ), striated muscle ( rhabdomyoma ) or fibrous tissue ( fibroma ).
 Lipoma (LP); (B) lipoma (HP); (C) angiolipoma (HP).")
Primary malignant tumours of soft tissue or sarcomas are rare and form a highly specialist area of practice. The approach to diagnosis is rather similar to that used in haematolymphoid pathology and typically requires careful correlation of the clinical and radiological findings with the histological appearance and pattern of immunohistochemical staining. Molecular studies may also be of value because many soft tissue tumours have characteristic non-random chromosomal rearrangements.
Classification of soft tissue tumours is complex and is currently based upon the World Health Organization (WHO) system. This uses a combination of clinical, histological, immunohistochemical and molecular features. In the past, sarcomas were often considered to arise from the tissue type that they resembled, e.g. liposarcomas were thought to develop due to malignant transformation of normal or benign adipose tissue. It is now accepted that most sarcomas do not arise from benign precursor lesions. Classification is therefore based upon the concept that tumour cells may differentiate so that they resemble normal tissues to a lesser or greater degree. This allows the description of groups of tumours that resemble tissues such as fat ( liposarcoma , see Fig. 22.16 ), smooth muscle ( leiomyosarcoma , illustrated in Fig. 7.6 ), striated muscle (rhabdomyosarcoma) , blood vessels ( angiosarcoma , illustrated in Fig. 11.12 ), nerves (malignant peripheral nerve sheath tumour) , fibrous tissue (fibrosarcoma) , etc.
.")
Some sarcomas lack any resemblance to normal tissues or structures but have sufficiently distinctive clinical, histological, immunohistochemical and molecular features to allow reliable identification. These are described within the WHO classification as tumours of uncertain histogenesis . The entity synovial sarcoma , despite its rather misleading name, is included within this group.
The term undifferentiated sarcoma is used for very aggressive and highly malignant tumours that do not resemble any normal tissues. By definition, these sarcomas show no immunohistochemical evidence of a particular pattern of differentiation and, as the name suggests, they are composed of highly pleomorphic malignant cells. An older term for these tumours still seen in some textbooks is malignant fibrous histiocytoma , based upon the supposed resemblance of the large and often multinucleate tumour cells to histiocytes. Pleomorphic sarcoma is one of the more common forms of soft tissue sarcoma in adults. An example is shown in Fig. 22.17 .
Soft tissue sarcomas occur over a wide age range and are one of the most common forms of malignancy in childhood. Benign soft tissue tumours usually occur in superficial sites whereas sarcomas are more likely to form deep-seated masses. Sarcomas are typically larger than benign tumours and, unlike carcinomas (which tend to spread through lymphatic channels to regional lymph nodes in the first instance), sarcomas typically metastasise via the bloodstream, giving rise to lung and bony metastases
Most malignant tumours, such as the common carcinomas, exhibit a wide range of genetic abnormalities, but these are rarely helpful in clinical diagnosis. In contrast, many soft tissue tumours exhibit highly characteristic non-random chromosomal rearrangements.
These are diagnostically useful in many cases and their recognition has also brought about advances in the understanding of tumour classification and in the development of some targeted drug therapies for the management of malignant disease (e.g. use of imatinib in treatment of gastrointestinal stromal tumours as discussed in Ch. 13 ).
Liposarcomas are interesting because molecular biology has contributed to the understanding of tumour classification. Myxoid liposarcomas typically exhibit a t(12;16) translocation, whilst well-differentiated and de-differentiated variants usually show evidence of a supernumerary giant ring or marker chromosome (formed of amplified genetic material from chromosome 12). If fresh tumour tissue is available, classical cytogenetic techniques can be used to demonstrate these abnormalities. Molecular methods have revealed that the area amplified on chromosome 12 typically includes the gene MDM2 (important in regulation of cell cycle). This MDM2 amplification can be detected using FISH techniques and the overexpressed protein can also be detected using immunohistochemical staining methods, which is helpful in routine diagnostic practice.
AM atypical mitosis GC tumour giant cells L lipoblast M mitotic figures
 MP; (B) HP.")
.")
 H&E (HP); (B) undecalcified resin section, Goldner trichrome stain (HP).")
 H&E (HP); (B) undecalcified resin section, Goldner’s trichrome (HP).")






 H&E (MP); (B) thin epoxy resin section, toluidine blue (HP).")




.")
 H&E (HP); (B) EM ×6000.")

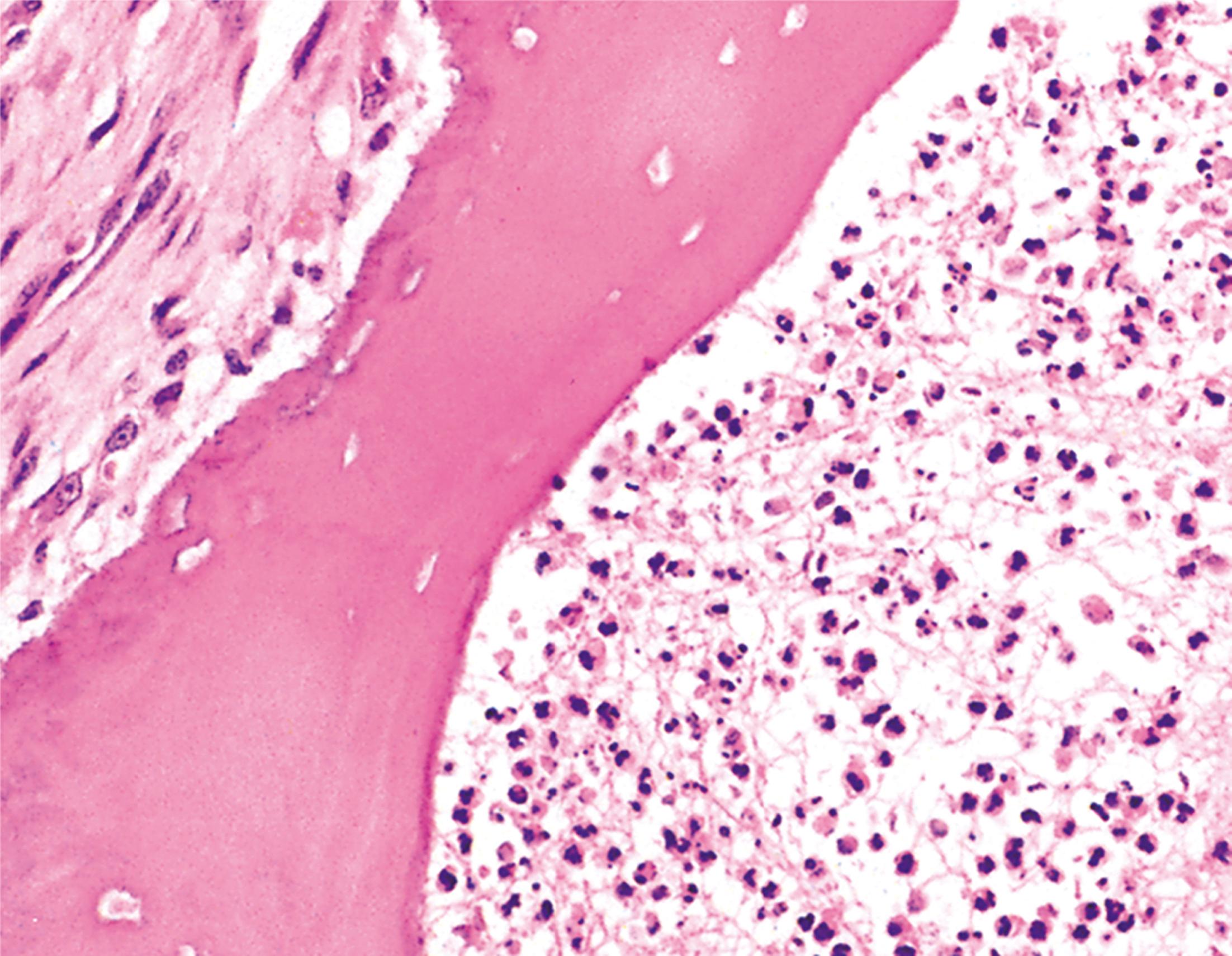
A 38-year-old gentleman with poorly controlled diabetes mellitus presents to the emergency department complaining of severe pain in his right foot. He has previously been treated for a long-standing ulcer over his right big toe. On examination, he is systemically unwell and febrile. There is swelling and redness extending along the medial border of the foot and it is exquisitely painful to touch. Following imaging, he undergoes a core biopsy from the first metatarsal bone, shown above. Which ONE of the following statements is INCORRECT? Choose ONE answer.
Become a Clinical Tree membership for Full access and enjoy Unlimited articles
If you are a member. Log in here
